Количественное определение содержания меди в порции мочи, которое позволяет оценить избыток или дефицит данного микроэлемента в организме.
Масс-спектрометрия с индуктивно-связанной плазмой.
Какой биоматериал можно использовать для исследования?
Разовую порцию мочи, суточную мочу.
Как правильно подготовиться к исследованию?
- Исключить из рациона алкоголь в течение 24 часов до исследования.
- Исключить (по согласованию с врачом) прием мочегонных препаратов в течение 48 часов до сбора мочи.
- Исключить физическое и эмоциональное перенапряжение во время сбора суточной мочи (в течение суток).
Общая информация об исследовании
Медь – жизненно важный микроэлемент, который входит в состав многих ферментов. Она необходима для синтеза гемоглобина, окислительно-восстановительных процессов, формирования соединительной ткани, синтеза пигмента меланина и функционирования нервной системы.
Медь содержат многие продукты питания: орехи, шоколад, грибы, зерно, некоторые морепродукты, печень, сухофрукты. В питьевую воду медь в небольших количествах попадает из медных труб, медной посуды. Данный микроэлемент всасывается в кишечнике, затем связывается с белками, превращаясь в нетоксическую форму, и транспортируется в печень. В печени медь накапливается и при необходимости доставляется в разные участки организма в комплексе с белком церулоплазмином. В кровотоке 95 % меди связано с церулоплазмином, а остальные 5 % – с альбумином. Только незначительная часть меди присутствует в крови в свободной форме. Избыток микроэлемента выделяется из организма с желчью и с мочой. В норме медь в моче выявляется в незначительном количестве. Дефицит или избыток меди возникает относительно редко, но имеет тяжелые последствия.
Болезнь Вильсона – Коновалова (гепатоцеребральная дистрофия) – генетическое заболевание с аутосомно-рецессивным типом наследования, которое приводит к избыточному накоплению меди во внутренних органах с преимущественным поражением печени и головного мозга. Одним из признаков заболевания являются кольца Кайзера – Флейшера вокруг радужки глаза, обусловленные отложениями меди. К симптомам болезни относятся анемия, тошнота, боль в животе, желтуха, дистония, тремор, двигательные и поведенческие расстройства. Схожая клиническая картина наблюдается при остром или хроническом отравлении медью на производстве, при загрязнении ею окружающей среды. Хронические заболевания печени также могут сопровождаться нарушением метаболизма меди.
Дефицит этого микроэлемента может возникнуть на фоне тяжелой мальабсорбции (нарушении всасывания питательных веществ из кишечника), например, при муковисцидозе, целиакии. Недостаточность меди в организме проявляется микроцитарной анемией, остеопорозом, нейтропенией. При тяжелом генетическом синдроме (болезни Менкеса), обусловленным врождённым нарушением всасывания меди из кишечника, у детей происходят глубокие нарушения развития нервной системы и истончение волос.
Несвоевременная диагностика состояний, сопровождающихся значительным нарушением содержания меди, может привести к тяжелым последствиям и даже к летальному исходу.
Для чего используется исследование?
- Для диагностики болезни Вильсона – Коновалова.
- Для диагностики острого или хронического отравления медью и медьсодержащими веществами.
- Для мониторинга лечения болезни Вилсона – Коновалова.
- Для диагностики дефицита или избытка меди в организме.
Когда назначается исследование?
- При подозрении на болезнь Вильсона – Коновалова (кольца Кайзера – Флейшера, патология печени и неврологические расстройства).
- При нарушении метаболизма меди у близких родственников.
- Во время лечения патологических состояний, сопровождающихся нарушением метаболизма меди.
- При подозрении на острое или хроническое отравление медью.
- При тяжелых заболеваниях печени.
Референсные значения: 2 — 80 мкг/л.
Причины повышения уровня меди в моче
- Болезнь Вильсона – Коновалова (значительное увеличение содержания меди в моче при сниженном уровне церулоплазмина и меди в крови).
- Острое или хроническое отравление медью (увеличение концентрации в моче и крови при нормальном уровне церулоплазмина).
- Билиарный цирроз.
- Хронический активный гепатит.
- Нефротический синдром.
- Гипоцерулоплазминемия.
- Болезнь Альцгеймера.
- Пеллагра.
- Проведение хелатной терапии (лечение отравлений медью и тяжелыми металлами, болезни Вильсона – Коновалова).
Причины снижения уровня меди в моче
- Дефицит меди в организме (на фоне мальабсорбции или неадекватного питания).
- Болезнь Менкеса.
Что может влиять на результат?
Ложное повышение меди в моче возможно:
- при употреблении витаминов, минеральных добавок и травяных препаратов, содержащих медь, за неделю до сдачи анализа;
- при беременности:
- при приеме карбамазепина, фенобарбитала, эстрогенов и пероральных контрацептивов.
- Для правильной интерпретации результатов необходимо учитывать содержание церулоплазмина и меди в крови.
Кто назначает исследование?
Терапевт, гепатолог, невролог.
- Назаренко Г. И., Кишкун А. Клиническая оценка результатов лабораторных исследований. – М.: Медицина, 2000. – С. 234-235.
- Wilson D. McGraw-Hill Manual of Laboratory and Diagnostic Tests 1st Ed. Normal, Illinois, 2007: p. 186-187.
источник
Болезнь Вильсона-Коновалова — (гепатоцеллюлярная дистрофия, болезнь Вильсона, гепатолентикулярная дегенерация) — редкое наследственное заболевание, наследуемое по аутосомно-рецессивному типу, проявляющееся преимущественно в молодом возрасте и характеризующееся избыточным накоплением меди в организме.
У пациентов медь накапливается в печени и мозге, а также в почках, роговице. Подробнее: Медь .
Болезнь Вильсона-Коновалова является причиной 15-20% всех болезней печени у детей.
Болезнь проявляется признаками поражения печени (часто развиваются цирроз печени и печеночная недостаточность ), нейропсихическими нарушениями, сочетанием указанных проявлений.
Патогномоничным симптомом для болезни Вильсона-Коновалова является обнаружение желто-коричневого кольца по периферии роговицы (кольцо Кайзера-Флейшера).
Диагноз заболевания устанавливается на основе физикального исследования, результатах лабораторных анализов, подтверждающих наличие нарушения метаболизма меди в организме (снижение уровня церулоплазмина в крови, повышение суточной экскреции меди с мочой); данных визуализирующих методов (УЗИ, КТ и МРТ ), при которых выявляются гепато- и спленомегалия, дегенерация базальных ганглиев головного мозга. В биоптатах печени обнаруживается повышенное содержание меди. Пациенту и его ближайшим родственникам проводится генетическое тестирование.
Лечение направлено на ограничение поступления меди в организм и уменьшение его содержания за счет назначения хелирующих препаратов ( D-пеницилламина , триентина).
- Классификация болезни Вильсона-Коновалова
В соответствии с клинической симптоматикой выделяются три формы заболевания:
- Болезнь Вильсона-Коновалова протекающая с преимущественным поражением ЦНС.
- Смешанная форма болезни Вильсона-Коновалова.
- Эпидемиология болезни Вильсона-Коновалова
В последние годы наблюдается тенденция к увеличению числа диагностируемых случаев болезни Вильсона-Коновалова. Распространенность заболевания в среднем — 30 случаев на 1 млн. человек.
В мире заболевание регистрируется с частотой 1: 35-100 тыс. новорожденных (уже насчитывается 10-30 млн. больных); носительство патологического гена отмечается в 0,56% случаев. В США частота выявления болезни Вильсона составляет 1:30 тыс. населения; носители мутантного гена (расположен на 13 хромосоме) обнаруживаются с частотой 1:90.
Высокая заболеваемость отмечается в регионах, где существуют близкородственные браки (Иран, Йемен, Ирландия), а также в Японии и на острове Сардиния. Так, в Японии болезнь Вильсона-Коновалова диагностируется с частотой 1:30 тыс.; для сравнения в Австралии — 1:100 тыс. населения.
Болезнь Вильсона-Коновалова встречается одинаково часто как у мужчин, так и у женщин.
Болезнь манифестирует в возрасте 8-16 лет, однако неврологические симптомы появляются только к 19-20 годам. У детей младше 5 лет проявления болезни Вильсона нередко могут отсутствовать, хотя заболевание иногда диагностируется как у пациентов в возрасте до 3 лет, так и у людей, которым уже за 50.
Без лечения болезнь Вильсона-Коновалова приводит к летальному исходу (примерно в возрасте 30 лет) в результате печеночной , почечной недостаточности, а также геморрагических осложнений.
Показатель смертности среди больных с возникшей фульминантной печеночной недостаточностью составляет 70%. Течение болезни Вильсона-Коновалова с развитием фульминантной печеночной недостаточности чаще наблюдается у женщин, чем у мужчин (4:1).
В 1883 г. C. Westphal и A. Strumpell описали сочетающееся с циррозом печени хроническое прогрессирующее заболевание нервной системы, назвав его псевдосклерозом. В 1912 г. S. Wilson опубликовал подробное описание клинической картины болезни, а саму болезнь назвал прогрессирующей лентикулярной дегенерацией. H.Hall в 1921 г. показал, что за названиями «псевдосклероз» и «гепатолентикулярная дегенерация» скрывается одна и та же нозология и ввел термин «болезнь Вильсона».
В 1953 г. Bearn, проведя анализ 30 семей, где были пациенты с болезнью Вильсона, установил аутосомно-рецессивный тип наследования этого заболевания.
В 1956 г. Walshe продемонстрировал хелирующий эффект препарата D-пеницилламина . В 1974 г. Frommer привел доказательства нарушения процесса билиарной экскреции меди при болезни Вильсона-Коновалова.
Мутантный ген (ATP7B), детерминирующий развитие этого заболевания и расположенный на 13 хромосоме (локус13q14-q21), был открыт Frydman и др. в 1985 г.
В дальнейшем было установлено, что этот ген кодирует кодирует белок, ответственный за внутриклеточный транспорт ионов меди (транспортирующий медь АТФазный протеин Р-типа). Медь — важный микроэлемент, так как он входит в состав целого ряда ферментов в организме. Но избыток меди приводит к цитотоксическим эффектам, которые опосредованы окислительными повреждениями клеточных мембран, дестабилизацией ядерной ДНК, разрушением лизосом.
В настоящее время идентифицировано более 200 мутаций гена ATP7B, которые приводят к нарушениям билиарной экскреции меди и к накоплению этого микроэлемента сначала в печени, а затем и в других органах и тканях (ЦНС, почках, сердце, костно-суставной системе). В результате возникает токсическое поражение этих органов и нарушение их функций.
По рекомендации ВОЗ суточная потребность в меди для взрослых составляет 1,5 мг. Содержание меди в обычной диете составляет 2-5 мг в день.
К продуктам с высоким содержанием меди относятся: баранина, свинина, мясо фазана, уток, гусей; кальмары, семга, субпродукты (печень, почки, сердце), морепродукты (устрицы, креветки, крабы, лобстер, морские гребешки, мидии), соевые продукты, орехи, грибы, сухофрукты (изюм, финики, чернослив), некоторые фрукты (авокадо), фасоль, горох, чечевица, пшено, ячмень, ржаной хлеб, свежий картофель, молочный шоколад, какао, минеральная вода.
Богатые источники меди содержат 0,3–2 мг/100 г продукта. Это: морепродукты, орехи, семена (включая порошок какао), бобы, отруби, зародышевые части зерен, печень и мясо.
Считается также, что 1 л питьевой воды содержит примерно 1 мг меди.
В организм медь поступает в основном с пищей. В желудочно-кишечном тракте абсорбируется до 95% поступившей в организм меди (причем в желудке ее максимальное количество), затем в двенадцатиперстной кишке, тощей и подвздошной кишке. Лучше всего организмом усваивается двухвалентная медь. В крови медь связывается с сывороточным альбумином (12-17%), аминокислотами — гистидином, треонином, глутамином (10-15%), транспортным белком транскуприном (12-14%) и церулоплазмином (до 60-65%). Небольшая часть меди ( 100-1000
Суточная экскреция меди с мочой при проведении пеницилламинового теста (прием 500 мг пеницилламина) мкг/сут Повышается незначительно >1500 Концентрация меди в ткани печени мкг/г 20-50 >250 (до 3000) Включение изотопа меди (64Cu или 67Cu) в церулоплазмин — Высокий уровень включения изотопа меди в церулоплазмин; второй пик через 48 часов. Не происходит включение изотопа меди в церулоплазмин; нет второго пика через 48 часов.
Критериями диагностики болезни Вильсона-Коновалова являются:
- Обнаружение кольца Кайзера-Флейшера.
- Снижение содержания церулоплазмина сыворотки крови (менее 20 мг/дл).
- Снижение содержания меди в сыворотке крови (менее 12 мкг/дл).
- Повышение экскреции меди с мочой (более 100 мкг/сут).
- Положительные результаты пеницилламинового теста.
- Повышенное содержание меди в ткани печени (более 250 мкг/г сухого вещества).
- Отсутствие включения изотопа меди в церулоплазмин.
У больного с нейропсихическими симптомами (или другими проявлениями, позволяющими заподозрить болезнь Вильсона-Коновалова) наличие кольца Кайзера-Флейшера и снижение содержания церулоплазмина сыворотки (менее 20 мг/дл) будут свидетельствовать в пользу болезни Вильсона-Коновалова.
Если у пациента имеются признаки хронического заболевания печени, но нет кольца Кайзера-Флейшера, то для установления диагноза болезни Вильсона-Коновалова достаточно получить доказательства повышенного содержания меди в ткани печени (более 250 мкг/г сухого вещества) и снижения содержания церулоплазмина в сыворотке крови.
Дифференциальный диагноз болезни Вильсона-Коновалова необходимо проводить со следующими заболеваниями и клиническими синдромами:
- Болезнь Паркинсона.
- Паркинсонизм, вызванный приемом лекарственных препаратов.
К паркинсонизму может приводить применение следующих лекарственных средств: нейролептиков — например, хлорпромазин ( Аминазин ); антиэметиков: например, метоклопрамид ( Церукал ); антигипертензивных средств: верапамил ( Изоптин , Феноптин ); метилдопы ( Допегит ), резерпина ; флунаризина (Сибелиум, Флунар); ловастатина ( Холетар , Кардиостатин ); амиодарона ( Кордарон ).
Паркинсонизм, вызванный воздействием токсических веществ.
Паркинсонизм может развиваться при контакте с монооксидом углерода; при отравлении цианидами; при отравлении магнием; при контакте с промышленными химикатами: дисульфидом углерода, метанолом, н-гексаном, дикватом (гербицид), разбавителями лаков.
Паркинсонизм, наблюдающийся при других патологических состояниях.
В эту группу входят случаи возникновения паркинсонизма при сосудистых и структурных аномалиях головного мозга; гидроцефалии; метаболических нарушениях (при гипертиреоидизме, гипопаратиреоидизме); гемиатрофии; травмах головного мозга; энцефалитах.
- Сенильная каротинемия.
- Хронический активный гепатит.
- Хронический холестаз.
- Хроническая желтуха .
- Множественная миелома.
- Первичный билиарный цирроз печени .
- Трипаносомоз.
источник
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Клинические протоколы МЗ РК — 2017

Одобрен объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения Республики Казахстан
от «28» ноября 2017 года
Протокол №33
Болезнь Вильсона-Коновалова (синонимы гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – тяжелое прогрессирующее наследственное заболевание, передающееся по аутосомно-рецессивному типу, в основе которого лежит нарушение экскреции меди из организма, приводящее к избыточному накоплению этого микроэлемента в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (преимущественно подкорковых ядер).
NB! Причиной возникновения БВК являются мутации гена ATP7B, который локализован на 13 хромосоме в локусе 13q14.3 и кодирует медь транспортирующую АТФ-азу Р-типа – ATP7B.
| МКБ-10 | |
| Код | Название |
| E83.0 | Нарушение обмена меди (Болезнь Вильсона-Коновалова) |
Дата разработки/пересмотра протокола: 2017 год.
Сокращения, используемые в протоколе:
| анти-LC | антитела к цитозольному антигену печени |
| анти-LKM | антитела к микросомам печени и почек |
| анти-LP | антитела к белкам печени и поджелудочной железы |
| анти-SLA | антитела к растворимым печеночным антигенам |
| БВК | болезнь Вильсона-Коновалова |
| БХАК | биохимический анализ крови |
| ГГТП | Гамма-глютамилтранспептидаза |
| ГЛД | гепатолентикулярная дегенерация |
| КТ | компьютерная томография |
| МРТ | магниторезонансная томография |
| ОАК | общий анализ крови |
| ОАМ | общий анализ мочи |
| СОЭ | скорость оседания эритроцитов |
| СРБ | С-реактивный протеин |
| УЗИ | ультразвуковое исследование |
| ЩФ | Щелочная фосфотаза |
| ЭКГ | электрокардиограмма |
| ЭНМГ | электронейромиография |
| ЭРХПГ | Эндоскопическая ретроградная холангиопанкреатография |
| ЭЭГ | электроэнцефалография |
| ANA | антинуклеарные антитела |
| IgG | иммуноглобулин G |
| АNCА | антитела к цитоплазме нейтрофилов |
| АМА | антимитохондриальные антитела |
Пользователи протокола: врачи общей практики, педиатры, детские гастроэнтерологи, детские неврологи.
Категория пациентов: дети.
Шкала уровня доказательности:
| A | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| B | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| C | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |

Клиническая картина гепатолентикулярной дегенерации характеризуется большим полиморфизмом в отношении как неврологических, так и соматических проявлений. Этот полиморфизм отражен в различных классификациях заболевания.
Формы болезни Вильсона [3]:
· Бессимптомная форма;
· Печеночная форма;
· Церебральная форма;
· Смешанная форма.
В зависимости от вовлечения в патологический процесс печени и центральной нервной системы и характера экстрапирамидной симптоматики, распознают 5 форм гепато-церебральной дистрофии [11]:
· Брюшная (абдоминальная) форма – манифестирует в возрасте от 5 до 17 лет и характеризуется различными вариантами поражения печени, нередко принимающими злокачественное «галопирующее» течение, приводящее к смерти раньше появления симптомов со стороны нервной системы. Её продолжительность от нескольких месяцев до 3-5 лет.
· Ригидно-аритмогиперкинетическая, или ранняя форма отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
· Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возраста, протекает медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжѐлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
· Дрожательная форма начинается в возрасте 20-30 лет, протекает довольно медленно (10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжѐлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
· Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепатоцеребральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжѐлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-6,10,19,20]: на БВ должны обследоваться дети в возрасте от 2 до 18 лет, имеющих необъяснимое повышение сывороточных аминотрансфераз, проявления фульминантной печеночной недостаточности, хронического гепатита, цирроза печени, неврологические нарушения неустановленной этиологии, Кумбс-негативную гемолитическую анемию, отягощенный семейный анамнез по БВ. Диагностика БВ базируется на комбинации клинических симптомов, данных лабораторного обследования и молекулярно-генетического тестирования.
Диагностические критерии [1-4]
Жалобы:
· боли в животе различной локализации;
· изменение цвета кожи;
· носовые кровотечения;
· тремор и непроизвольные движения;
· слюнотечение, дизартрия, нарушение глотания;
· мигренеподобные головные боли;
· бессонница;
· депрессия;
· невротическое поведение;
· изменения личности;
· психоз.
Анамнез: Первичная манифестация БВ может протекать в виде острого фульминантного гепатита, проявляющегося коагулопатией, энцефалопатией, Кумбс-негативной гемолитической анемией, печеночноклеточной и почечной недостаточностью, с выявлением значительного превышения меди в сыворотке крови и моче.
NB! Обратить внимание на возраст начала проявлений заболевания у пациента: до 5 летнего возраста проявления болезни Вильсона-Коновалова, как правило, отсутствуют. Болезнь манифестирует в возрасте 8-16 лет (хотя практически с рождения отмечается повышенная активность печеночных аминотрансфераз). Необходимо уточнить о наличии заболеваний печени и нейропсихических нарушений у ближайших родственников больного (стеатоза, гепатитов, цирроза печени, печеночной недостаточности).
NB! Первые симптомы заболевания начинается с симптомов поражения печени (в 42% случаев). Примерно у 25% пациентов заболевание начинается остро, с развития желтухи, астенического синдрома, анорексии, повышения температуры. Болезнь Вильсона-Коновалова может клинически протекать по типу аутоиммунного гепатита, с повышением уровня сывороточных иммуноглобулинов и неспецифических аутоантител, в связи с чем необходимо исключать данное заболевание и у больных с аутоиммунным гепатитом. Неврологическая и психическая симптоматика наблюдается у 10% больных. У 15% пациентов болезнь Вильсона-Коновалова манифестирует гематологическими синдромами (прежде всего гемолитической анемией). Кольцо Кайзера-Флейшера не выявляется у детей до 5 лет.
Клинические признаки болезни отражены в таблице 1.
Таблица 1 — Клинические признаки болезни Вильсона.
| Проявления болезни Вильсона-Коновалова | Симптомы |
| Поражение печени | Бессимптомная гепатомегалия |
| Изолированная спленомегалия | |
| Стеатогепатит | |
| Острый (фульминантный) гепатит | |
| Аутоиммуноподобный гепатит | |
| Цирроз печени | |
| Поражение ЦНС | Двигательные нарушения (тремор, непроизвольные движения) |
| Слюнотечение, дизартрия | |
| Ригидная дистония | |
| Псевдобульбарный синдром | |
| Вегетососудистая дистония | |
| Мигренеподобные головные боли | |
| Бессоница | |
| Дистонические атаки | |
| Психиатрические симптомы | Депрессия |
| Невротическое поведение | |
| Изменения личности | |
| Психоз | |
| Другие системы | Офтальмология: кольца Кайзера-Флейшера, «медная» катаракта |
| Гемолитическая анемия | |
| Патология почек: аминоацидурия, нефролитиаз | |
| Патология скелета: ранний остеопороз, артрит | |
| Поражение сердца: кардиомиопатия, нарушения ритма | |
| Панкреатит, желчекаменная болезнь | |
| Гипопаратиреодизм, гигантизм |
Физикальное обследование [1-5]:
необходимо оценить наличие:
· смуглого («медный») цвета кожи;
· желтушности склер;
· незначительной или умеренной гепатомегалии;
· спленомегалии;
· неврологических нарушений и психических расстройств в виде непроизвольных движений в мышцах торса и конечностей;
· мигренеподобной головной боли;
· скованности в мышцах;
· эмоциональной лабильности;
· агрессивности.
Лабораторные исследования [1-6]:
· общий анализ крови: лейкопения, нормохромная анемия, тромбоцитопения, ретикулоцитоз, ускоренная СОЭ.
· общий анализ мочи: при поражении почек можно обнаружить микрогематурию, незначительную протеинурию, гиперкальциурию.
· суточная экскреция мочи: гиперкупренилурия, признаки развившейся тубулопатии с признаками: глюкозурией, аминоацидурией, фосфатурией, уратурией, протеинурией.
· биохимический анализ крови: снижение церулоплазмина и общей меди, увеличение уровней свободной меди (таблица 1), аминотрасфераз (в 1,5-50 раз); билирубин повышен более чем в 2 раза, преимущественно за счет прямой фракции; уровень щелочной фосфатазы обычно повышен; может быть повышена активность гаммаглютамилтранспептидазы (ГГТП); гипоальбуминемия.
· коагулограмма: снижение протромбинового индекса, гипофибриногенемия, снижение тромбинового времени.
· пеницилламиновый тест: необходимо исследовать мочу, собранную сразу после приема 500 мг пеницилламина и через 12 часов. У пациентов с болезнью Вильсона-Коновалова суточная экскреция меди будет повышаться до более 1500 мкг/дл/сут (норма
| Показатель | Норма | ГЛД |
| Церулоплазмин, мг/дл | 17–40 | |
| Общая медь в сыворотке крови, мкмоль/л | 12–32 | |
| Свободная медь в сыворотке крови, мкг/дл | 5–12 | > 50 |
| Суточная экскреция меди с мочой, мкг/сут | > 100–1000 | |
| Суточная экскреция меди с мочой в пробе с Д-пеницилламином, мкг/сут | 600–800 | 1000–3000 |
| Количественное содержание меди в печени, мкг/г | 15–55 | > 250 |
Инструментальные исследования [1-6]:
· УЗИ печени и селезенки: позволяют выявить увеличение печени и реже селезенки, признаки портальной гипертензии и цирроза печени [5].
· ЭКГ – при поражении сердца можно выявить признаки гипертрофии левого или обоих желудочков, депрессию сегмента ST, инверсию зубца Т, различные виды нарушений ритма.
· ЭхоКГ – в ходе этого исследования можно выявить кардиомиопатию, гидроперикард.
· Электроэнцефалография – проводится пациентам с тяжелыми нарушениями со стороны ЦНС с эпилептическими приступами, регистрируется эпилептическая активность.
· Эзофагогастродуоденоскопия: на наличие варикозно расширенных вен пищевода и желудка.
· МРТ головного мозга: более информативно в диагностике, чем КТ головного мозга. Характерны билатеральные очаги пониженной плотности 3-15 мм в диаметре в области базальных ганглиев (хвостатое ядро, скорлупа и бледный шар), в таламусе, в области зубчатых ядер и коры мозжечка — симптом «морды гигантской панды» [19]. По мере прогрессирования процесса выявляются признаки диффузного атрофического процесса головного мозга с равномерным расширением субарахноидальных пространств и желудочковой системы [20]. Изменения МР-сигнала от структур головного мозга могут отсутствовать у 7-17% детей со смешанной формой болезни Вильсона при наличии неврологической симптоматики в виде тремора, дизартрии и изменения мышечного тонуса по экстрапирамидному типу [1-10].
· Пункционная биопсия печени: 1. для морфологического исследования биоптата печени, где выявляются дистрофические изменения клеток, некрозы, слабую воспалительную инфильтрацию и фиброз различной степени выраженности; 2. Определения концентрации меди в препарате печени — при ГЛД медь находится на уровне 1 000 мкг на 1 г сухого вещества печени. На доклинической стадии уровень печеночной меди не всегда превышает 250 мкг/г, у гетерозиготных носителей может быть в пределах 150–200 мкг/г. Нормальный уровень печеночной меди исключает ГЛД, тогда как повышенный подтверждает диагноз при наличии соответствующих клинических данных [1-7].
· Гистологическое исследование биоптата печени: морфологические изменения печени при болезни Вильсона не являются патогномоничными и включают в себя на ранних стадиях заболевания признаки жировой инфильтрации гепатоцитов (крупнокапельной и мелкокапельной), гликогеновой дегенерации ядер и фокальный гепатоцеллюлярный некроз, а также преобладание минимальной и низкой степени активности воспалительного процесса, нередко в сочетании с выраженными фибротическими изменениями. Могут быть изменения по типу аутоиммунного гепатита, вирусного, алкогольного и лекарственного поражения печени. По мере прогрессирования повреждений паренхимы, формируется фиброз и, впоследствии, цирроз печени. Обычно встречается гистологическая картина крупноузлового цирроза печени в исходе болезни Вильсона, однако, встречаются случаи и мелкоузлового цирроза.
Показания для консультации специалистов [1-4]:
· консультация офтальмолога – для выявления колец Кайзера-Флейшера, также на наличие катаракты у детей, с целью исключения других болезней накопления;
· консультация невропатолога – оценка неврологического статуса, нервно-психического статуса;
· консультация психиатра – диагностика психиатрических состояний;
· консультация психотерапевта – коррекция психологических проблем;
· консультация гастроэнтеролога – коррекция нарушений желудочно-кишечного тракта;
· консультация гематолога – при наличии симптомов гемолитической анемии, коагулопатии;
· консультация нефролога – при наличии патологии в анализах мочи;
· консультация сурдолога – определение остроты слуха;
· консультация физиотерапевта – определение методов физиотерапевтического лечения;
· консультация хирурга – при риске пищеводно-желудочных кровотечений, для выявления показаний к проведению трансплантации печени у детей с признаками цирроза печени (ЦП), печеночно-клеточной декомпенсацией;
· консультация инфекциониста – при наличии сопутствующего вирусного гепатита;
· консультация отоларинголога – при инфекциях верхних дыхательных путей.
Диагностический алгоритм: 
Рисунок 1- Алгоритм диагностики болезни Вильсона [3]. КФ – кольца Кайзера-Флейшера; Цер – церулоплазмин; 24-h Cu – суточная экскреция меди с мочой.
Ни один лабораторный тест (за исключением полного секвенирования патологического гена АТР7В) не обладает 100% чувствительностью и не обеспечивает 100% специфичность, таблица 3.
Таблица 3 — Тесты для диагностики болезни Вильсона.
| Тест | Характерные находки | Ложноотрицательный результат | Ложноположительный результат |
| Сывороточный церулоплазмин | Уменьшение на 50% относительно нормы | Нормальный уровень у пациентов с выраженным воспалением в печени. Завышение результата при иммунологических методах исследования. Беременность, прием эстрогенов. | Низкий уровень при: -мальабсорбции; -ацерулоплазминемии; -гетерозиготы |
| Суточная экскреция меди с мочой | >0,64мкмоль/24часа | Норма: -неправильный сбор мочи; -дети без вовлечения печени | Повышение: -гепатоцеллюлярный некроз; -холестаз; -контаминация |
| Свободная медь сыворотки | >1,6мкмоль/л | Норма при завышении уровня церулоплазмина иммунологическими методами | |
| Печеночная медь | >4мкмоль/г сухого вещества | В зависимости от места взятия материала: -активная болезнь печени; -узелки регенерации | Синдромы холестаза |
| Кольцо Кайзера-Флейшнера (при использовании щелевой лампы) | Наличие кольца | Отсутствует: -у 50% пациентов с печеночной формой; -у большинства асимптоматических сибсов | Первичный биллиарный цирроз |
Диагноз болезни Вильсона ставится на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа (таблица 4).
Таблица 4 — Лейпцигские балльные критерии диагностики болезни Вильсона [11]
| Типичные клинические симптомы и признаки | Другие тесты |
| Кольцо Кайзера-Флейшнера: Присутствует 2 Отсутствует 0 | Печеночная медь (при отсутствии холестаза) >5х верхней границы нормы (>4мкМ/г) 2 0,8-4мкМ/г 1 Норма ( |
| Неврологическая симптоматика или типичные изменения на МРТ головного мозга: Тяжелая 2 Средняя 1 Отсутствует 0 | Медь в моче (в отсутствии острого гепатита) Норма 0 1-2х верхней границы нормы 1 >2х верхней границы нормы 2 Норма, но увеличение >5х верхней границы нормы после пеницилламина 2 |
| Церулоплазмин сыворотки Норма (>0,2г/л) 0 0,1-0,2 г/л 1 | Анализ мутаций Обе хромосомы 4 На одной хромосоме 1 Не выявлено мутаций 0 |
| Кумбс-негативная гемолитическая анемия Присутствует 1 Отсутствует 0 | |
| Интерпретация результата | |
| 4 и более | Диагноз установлен |
| 3 | Диагноз возможен, необходимы дальнейшие исследования |
| 2 и менее | Диагноз маловероятен |
Дифференциальный диагноз и обоснование дополнительных исследований [11-20]
Таблица 5– Дифференциальный диагноз болезни Вильсона-Коновалова.
источник
Сайт предоставляет справочную информацию исключительно для ознакомления. Диагностику и лечение заболеваний нужно проходить под наблюдением специалиста. У всех препаратов имеются противопоказания. Консультация специалиста обязательна!
Печень – самый большой внутренний орган человека. Вес печени 1.2 – 1.5 килограммов (это примерно 2% массы тела). Печень расположена под диафрагмой в правой верхней части живота. В норме нижний край печени находится примерно на уровне последнего ребра (справа).
Печень разделяют на 4 доли: левую, правую, квадратную и хвостовую. Орган покрыт капсулой, которая содержит в себе обильную иннервацию.
Печень состоит из клеток (гепатоцитов). Она имеет необычное кровоснабжение. Обычно в сам орган (в любой другой, кроме печени) входит артерия, которая снабжает богатой кислородом кровью ткани, и выходит вена, которая выносит из органа кровь.
В печень, помимо артерии, входит воротная вена, а выходит печеночная вена. Кровь в воротную вену поступает из кишечника и других органов. Эта кровь полна токсических веществ. Она попадает в печень, где все токсические вещества нейтрализуются.
Желчевыводящая система печени. Желчь, образованная в печени, через желчные канальцы собирается в более крупные каналы, которые впоследствии образуют общий желчный проток. Далее желчь поступает в желчный пузырь.
 Функция депо
Функция депо
В печени происходит накопление (депонирование) гликогена, жирорастворимых витаминов (А, D, E, K).
Гликоген накапливается в печени под действием инсулина. В случае необходимости (снижение уровня глюкозы в крови) гликоген выходит из печени и под действием глюкагона превращается в глюкозу. Тем самым происходит поддержание сахара крови на одном уровне.
Участие во всех видах обмена веществ (особенно липидном и белковом).
Дезинтоксикационная функция
Печень — главный орган, обезвреживающий множество токсических веществ. Процесс дезинтоксикации осуществляется с помощью химических превращений токсических веществ в нетоксические соединения.
Токсины бывают эндогенного и экзогенного происхождения. Экзогенные токсины — это различные медицинские препараты и токсические продукты, образующиеся в кишечнике под действием бактерий. В кишечнике образуются такие токсические вещества, как индол, скатол, и главное — аммиак.
Аммиак печень обезвреживает до мочевины, а потом мочевина выделяется почками вместе с мочой. Также печень обезвреживает множество гормонов (глюкокортикоиды, альдостерон, инсулин, эстрогены и множество других гормонов) для поддержания необходимого гормонального равновесия. Печень обезвреживает токсины посредством окисления, ацетилирования.
Синтез белков крови
Печень синтезирует фибриноген, протромбин, альбумины.
Фибриноген – белок плазмы крови, который участвует в свертывании крови (фактор свертывания 1).
Протромбин – белок, который участвует в свертывании крови (фактор свертывания номер 2).
Альбумины – белки, участвующие в транспорте множества веществ. Например, одна молекула альбумина может одновременно переносить 25-50 молекул билирубина.
Секреторная функция
Печень секретирует желчь, которая необходима для пищеварения. Желчь активирует множество поджелудочных ферментов (трипсин, липаза). Также желчь участвует в расщеплении липидов (жиров).
С древних времен люди использовали лечебные свойства меди. Клеопатра, к примеру, носила медные браслеты для сохранения молодости кожи, а античные войны часто использовали медь как материал для брони. Считалось что воины, облаченные в такую броню, дольше не уставали, а их раны не гноились и быстрее заживали.
- Медь принимает участие в синтезе многих протеинов (белков) и ферментов, а также в процессе роста и развития организма.
- Медь участвует в превращении железа в гемоглобин. Также медь является частью ферментов, синтезирующих эритроциты и лейкоциты.
- Благодаря меди кровеносные сосуды сохраняют свою эластичность.
- С помощью меди транспортирующие системы успешно переносят железо из печени в необходимое место. Без меди этот транспорт невозможен.
- Медь участвует в синтезе коллагена (белок, обеспечивающий прочность и эластичность тканей), который участвует в создании каркаса скелетных костей.
- Соединяясь с аскорбиновой кислотой, медь участвует в поддержании иммунной системы в активном состоянии.
- Медь также необходима для нормальной работы фермента супероксиддисмутазы (мощный антиоксидант). С помощью данного фермента предотвращается преждевременное старение кожи. Также хочется отметить, что этот фермент входит в состав косметических средств против старения кожи.
- Медь стимулирует активность гормонов гипофиза. В присутствии меди активность инсулина в крови (гормон снижающий сахар крови) повышается.
- Медь необходима и для пищеварительной системы. Ученые установили, что она защищает пищеварительную систему от повреждения и воспаления. Некоторые ученые считают, что медь даже может заживить небольшие язвыжелудка.
Болезнь Вильсона — это наследственное заболевание, которое передается по аутосомно-рецесивному типу (оба родителя являются носителями аномального гена).
Причиной заболевания является мутация гена, отвечающего за синтез белка, который осуществляет транспорт меди (церулоплазмин). Этот ген имеет название AT- P7B, и он находится на длинном плече хромосомы под номером 13.
Ученые насчитали около 80 возможных вариантов мутаций данного гена. Самые опасные мутации — это те, которые вызывают его полное разрушение. Естественно, такие мутации приводят к заболеванию в очень тяжелой форме.
Что же происходит в организме из-за нарушения обмена меди?
Обменный дефект при болезни Вильсона состоит в невозможности поддержания баланса меди в организме.
В случае избытка медь накапливается в печени. В то же время блокируется процесс выделения микроэлемента с желчью, что еще больше увеличивает ее количество.
Постепенно избыток меди приводит к хронической интоксикации. Медь накапливается и в других органах и системах (нервная система).
Встречается чаще всего: 50-80% случаев. Развивается либо как хронический гепатит, либо как цирроз печени. Характерные симптомы заболевания:
Желтуха 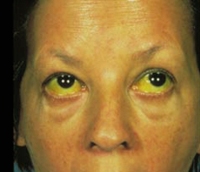 Желтуха – окрашивание кожных покровов и слизистых оболочек в желтушный цвет. Желтуха появляется из-за увеличения количества различных фракций билирубина в крови. Как правило, «пожелтение» начинается со слизистых оболочек (склера глаза, слизистая под языком, мягкое нёбо). Постепенно желтушность распространяется и на кожные покровы. В первую очередь желтеет кожа лица. Постепенно начинают менять цвет кожные покровы верхних, а затем нижних конечностей. Область живота меняет свой цвет в последнюю очередь. Если у пациента пожелтела вся кожа, тогда говорят о сильно выраженной желтухе. Выраженная желтуха всегда сопровождается сильной интоксикацией организма.
Желтуха – окрашивание кожных покровов и слизистых оболочек в желтушный цвет. Желтуха появляется из-за увеличения количества различных фракций билирубина в крови. Как правило, «пожелтение» начинается со слизистых оболочек (склера глаза, слизистая под языком, мягкое нёбо). Постепенно желтушность распространяется и на кожные покровы. В первую очередь желтеет кожа лица. Постепенно начинают менять цвет кожные покровы верхних, а затем нижних конечностей. Область живота меняет свой цвет в последнюю очередь. Если у пациента пожелтела вся кожа, тогда говорят о сильно выраженной желтухе. Выраженная желтуха всегда сопровождается сильной интоксикацией организма.
Существует три вида желтухи: печеночная, механическая, гемолитическая.
Механическая желтуха появляется при закупорке выводных желчных протоков. Цвет кожи приобретает зелено-желтый оттенок. Также характерным симптомом является кожный зуд. Желтуха очень сильно выражена.
Гемолитическая желтуха появляется при повышенном разрушении эритроцитов (красных телец крови). Цвет кожи при этом становится бледно-лимонным. Обычно данный вид желтухи не интенсивен. Кожный зуд отсутствует.
Печеночная желтуха
Данная форма желтухи проявляется при болезни Вильсона. Печеночная желтуха характеризуется поражением клеток печени, вырабатывающих желчь, что ведет к попаданию билирубина в кровь. Цвет кожи при данной форме желтухи становится желто-оранжевым. Интенсивность печеночной желтухи — умеренная. Кожный зуд появляется редко. Характерными признаками печеночной желтухи служат моча темного цвета (коньячного) и кал белого (бесцветный).
При печеночной желтухе в крови повышается связанный билирубин. Этот билирубин легко выделяется с мочой. Именно поэтому моча приобретает коньячный цвет.
Как было описано выше, разрушаются клетки печени, что ведет к снижению выработки желчи. Именно метаболиты желчных кислот окрашивают кал в нормальный цвет. А раз желчь не вырабатывается, поэтому и кал бесцветный.
Асцит
Асцит – скопление в брюшной полости жидкости. Этот симптом появляется в данном случае при значительном поражении печени. Жидкость, похожая по составу на плазму крови, скапливаясь в брюшной полости, давит на все органы, которые там расположены.
У пациента с выраженным асцитом увеличен в размере живот. Причем при смене положения тела увеличенный живот тоже меняет свое положение. Жидкость двигается под действием силы притяжения земли. Жидкость всегда двигается в место, наиболее приближенное к поверхности земли. Поэтому пациентов с асцитом легко отличить от пациентов с выраженным ожирением (при ожирении живот не меняет свое положение).
Отёки
Отёки появляются чаще всего на ногах. При поражении почек отеки возникают под глазами.
Кровотечения
Кровотечения происходят из-за нарушения свертываемости крови. Печень вырабатывает различные факторы свертывания. При болезни Вильсона клетки печени разрушаются, поэтому печень не способна вырабатывать факторы свертывания крови.
Аменорея
Аменорея – это отсутствие у женщин менструальных циклов. Печень помимо всего еще и инактивирует различные гормоны. В норме печень инактивирует избыток эстрогенов. В данном случае печень не выполняет этой функции, тем самым вызывая аменорею.
 Характерными симптомами заболевания являются:
Характерными симптомами заболевания являются:
1. Тремор рук, головы. Тремор – непроизвольные быстрые движения рук или головы. Для тремора характерна ритмичность, небольшой объем (размах) движений.
Тремор может быть постоянным или периодическим (например, отсутствие тремора во время сна).
2. Гримасничанье – преувеличенные быстро меняющиеся движения лицевых мышц, возникающие непроизвольно.
3. Нарушения почерка. Почерк становится неровным, неразборчивым. При написании какого-либо текста наблюдается неодинаковые по размеру и расположению буквы в слове (одни буквы меньше — другие больше, одни выше — другие ниже).
4. Дизартрия – расстройство речи, при котором происходит нарушение произношения звуков, слов. Речь становится непонятной, «говорит, как с кашей во рту». Данное расстройство возникает вследствие нарушения иннервации органов, участвующих в создании речи (мягкого нёба, языка, губ).
5. Более поздними симптомами являются: контрактура при сгибании и мышечная ригидность.
Контрактура — ограничение пассивных и движений в различных суставах. В данном случае ограничивается разгибательные движения в суставах рук и ног.
Мышечная ригидность – повышенный тонус мышц. Из-за увеличенного тонуса различных групп мышц происходит увеличение сопротивления при осуществлении движений (труднее совершить то или иное движение).
6. Нарушение психики встречается в 20% случаев. Среди нарушений можно отметить психоз, депрессию.
Психоз – психическое расстройство, проявляющееся крайне неадекватным поведением, с присутствием таких явлений, как галлюцинации и бред.
Депрессия – расстройство, при котором происходит значительное и стабильное снижение настроения, утрата способности радоваться, потеря интереса к жизни, двигательная заторможенность.
Симптомом, характерным для обеих форм болезни Вильсона, служит Кольцо Кайзера — Флейшера. Он встречается в 50-62% случаев.
Кольцо Кайзера – Флейшера – это кольцо коричневатого цвета, которое расположено на наружном крае роговицы глаза. Оно появляется из-за накопления в роговице меди.
Интересно, что хорошо выраженное кольцо можно увидеть невооруженным глазом. Если интенсивность кольца невелика, чтобы его определить, необходимо специальное оборудование.
1. Изменения со стороны почек встречается в 10% случаев. Они случаются из-за накопления меди в клетках почек. Чрезмерное накопление меди ведет к разрушению клеток почки, в результате чего появляются следующие симптомы:
- Гематурия – присутствие в моче элементов крови (эритроцитов). В норме в моче нет крови.
- Гликозурия – присутствие в моче глюкозы. В норме в моче не должно быть глюкозы.
2. Внутрисосудистый гемолиз – разрушение эритроцитов в полости сосудов. Встречается в 10-15:% случаев. Гемолиз в конечном итоге приводит к анемии (снижение количества эритроцитов и гемоглобина в крови).
3. Поражение костной системы — встречается в 20% случаев. Одним из главных проявлений является остеопороз.
Остеопороз – заболевание, при котором снижается плотность, и нарушается структура костей. Вследствие остеопороза повышается ломкость костей, что в свою очередь ведет к частым переломам.
Болезнь Вильсона можно разделить на 2 стадии.
Латентная стадия – стадия заболевания, в которой отсутствуют симптомы, но присутствуют изменения (накопление меди) на уровне печени. Продолжительность этой стадии — в среднем от 5 до 7 лет.
В случае, если лечение не было назначено, заболевание постепенно переходит в стадию клинических проявлений (симптомы).
Если после появлений симптомов правильное лечение не назначается, возможны осложнения.
Беседа у врача
Доктор расспросит вас о жалобах. Особенно углубленно расспросит о том, было ли похожее заболевание у отца, матери или других родственников.
Осмотр
Как правило, осмотр при болезни Вильсона начинают с глаз. Доктор ищет кольцо Кайзера – Флейшера.
Прощупывание (пальпация) печени
При пальпации печень увеличена, консистенция, как правило, слегка уплотненная.
 Общий анализ крови
Общий анализ крови
В данном случае общий анализ крови не подтверждает присутствие болезни. Поэтому он осуществляется только для исключения осложнений (анемия вследствие гемолиза).
Биохимический анализ крови
АЛАТ и АСАТ увеличены.
Дозирование церулоплазмина (белок-переносчик меди). При болезни Вильсона оно падает ниже 20 мг/децилитр. Специфичность (характерность для данного заболевания) этого теста — 90%.
Определение количества меди в крови. Увеличение количества меди — выше 1500 мг на литр.
Анализ мочи на присутствие меди
В моче обнаруживается медь (купруурия) от 100 до 1000 микрограмм в сутки.
Биопсия печени
Биопсия — это взятие кусочка ткани или органа для исследования. Биопсию печени производят с помощью специальной тонкой иглы. С помощью иглы берут кусочек ткани, затем изучают его под микроскопом или делают различные лабораторные анализы.
В данном случае печень исследуют на содержание в ней меди. В норме в печени находится 50-100 микрограмм меди на 1 грамм высушенной ткани.
При болезни Вильсона количество меди превышает 250 микрограмм на 1 грамм высушенной ткани печени.
Меченая медь
Существует метод с использованием меченой меди. Микроэлемент вводят в организм, а затем наблюдают за его накоплением и передвижением (транспортом).
Генетическое исследование
Данный метод предполагает использование ДНК–маркеров, которые помогают установить диагноз с высокой точностью. Недостатками данного метода являются его высокая стоимость, а также большая вариабельность мутаций гена болезни Вильсона.
Фульминантная форма гепатита
Эта форма сильно отличается от тяжелой формы гепатита. При фульминантной форме происходит массовый некроз (разрушение) клеток печени. Обычно клетки печени очень хорошо регенерируют (восстанавливаются) даже при тяжелой форме гепатита. При фулминантной форме регенерация отсутствует. Характерный симптом данного гепатита — это «тающая печень» (печень быстро уменьшается в размерах).
Это осложнение крайне тяжелое, так как очень часто приводит к смертельному исходу.
Цирроз печени
Цирроз – замещение нормальной (характерной для того или иного органа) ткани на соединительную (фиброзную) ткань. Цирроз печени приводит к постепенному нарушению ее функций. Особенно сильно нарушаются функция дезинтоксикации и синтеза необходимых белков.
Гемолиз
Гемолиз – повышенное разрушение красных телец крови (эритроцитов). Приводит к появлению анемии (снижение количества эритроцитов и гемоглобина в крови). Анемия, в свою очередь, приводит к гипоксии (недостаточное поступление кислорода) органов и тканей. Гипоксия приводит к нарушению метаболизма, а затем к нарушению работы органов.
Образование камней в почках
Это осложнение происходит из-за нарушения баланса солей в организме, и из-за накопления в почках меди. Камни мешают нормальной работе почек (ухудшают процесс фильтрации и усложняют процесс выделения мочи).
Рекомендации по питанию:
- в рационе должен присутствовать легкоусвояемый белок;
- количество жиров в рационе должно быть снижено;
- количество углеводов в диете не должно превышать физиологические нормы;
- кулинарная обработка должна быть щадящей (вареная пища, пища на пару, пища в печеном виде);
- пища должна быть измельченной;
- в диету должны быть включены продукты, содержащие пищевые волокна.
Главное условие диеты при болезни Вильсона — исключение из рациона продуктов, содержащих медь.
Продукты, содержащие медь:
- баранина;
- мясо курицы;
- утка;
- колбаса;
- ракообразные;
- щавель;
- сладкий перец;
- бобовые;
- орехи;
- шоколад, какао, кофе;
- мёд;
- минеральная вода;
- грибы;
- печень.
Следует помнить, что диету необходимо держать на протяжении всей жизни!
 Для лечения болезни Вильсона используют препараты, связывающие и выводящие медь из организма.
Для лечения болезни Вильсона используют препараты, связывающие и выводящие медь из организма.
Д–пеницилламин – препарат, который связывается с медью, образуя водорастворимые соединения. Эти соединения впоследствии выделяются с мочой. Д–пеницилламин применяют в дозе 150 миллиграмм в сутки (15 дней), затем 300 миллиграмм в день (15 дней), потом 600 миллиграмм (15 дней), и наконец, 1000-2000 миллиграмм — доза, которая принимается каждый день в течение всей жизни.
В случае появления побочных эффектов на Д-пеницилламин, назначают другой препарат. Триентин применяется в дозировке 750-2000 миллиграмм в сутки.
Также возможно применение солей цинка (ацетат цинка, сульфат цинка). Эти препараты применяются в дозировке 100-150 миллиграмм цинка в день.
На данный момент самая эффективная схема лечения болезни Вильсона — это комбинирование препаратов цинка и пеницилламина.
В случае развития фулминантного гепатита (осложнение, описанное выше) единственный способ лечения — это пересадка печени.
В случае неврологических симптомов назначают общеукрепляющие препараты.
В случае тремора рук и гримасничания назначают лоразепам или клоназепам.
В случае депрессии назначают различные антидепрессанты.
Чтобы защитить печень назначают гепатопротекторы, например силимарин.
Лечение осуществляется под обязательным контролем лабораторных исследований на наличие меди.
В случае редких психических отклонений (психоз) рекомендуется консультация психиатра.
Первичная профилактика
Это метод профилактики, который предполагает установления диагноза пренатально (до рождения ребенка). Диагноз может быть поставлен с помощью консультации генетика.
Вторичная профилактика
Предполагает назначение лечения до появления симптомов заболевания, а также проверка всех членов семьи на наличие данного заболевания. Для этого необходимо обследование больного, а также сдача анализов на наличие меди в крови и моче. Дети младше 6 лет должны быть повторно обследованы в течение 5-10 лет.
Пациент, принимающий лечение, должен находиться на учете у врача-гастроэнтеролога постоянно.
Автор: Пашков М.К. Координатор проекта по контенту.
источник
| БОЛЕЗНЬ ВИЛЬСОНА — КОНОВАЛОВА (Гепатолентикулярная дегенерация) |
Это тяжелое прогрессирующее экстрапирамидное заболевание, постоянным и характерным признаком которого является поражение печени.
Заболевание наследственное, передается по аутосомно-рецессивному типу. С помощью метода рекомбинации ДНК получены данные, свидетельствующие о локализации патологического процесса, ответственного за развитие болезни, на хромосоме 13.
Ведущую роль в патогенезе играет патология обмена меди, вследствие нарушения синтеза фермента церулоплазмина, которые приводят к отложению избыточного количества меди в различных органах и тканях (преимущественно в печени, мозге, почках и роговице) и развитию вследствие этого клинических симптомов болезни.
Поражение печени имеет характер крупноузлового или смешанного (крупноузлового и мелкоузлового) цирроза.
Развитие цирроза печени может проходить через стадию хронического активного гепатита.
В почках медь откладывается главным образом в проксимальных отделах почечных канальцев, что приводит к нарушению реабсорбции целого ряда важных для организма соединений и может сопровождаться определенными метаболическими сдвигами.
В результате отложения меди в десцеметовой мембране роговицы образуется роговичное кольцо Кайзера — Флейшера, которое является патогномоничным признаком болезни.
В мозге медь преимущественно откладывается в базальных ганглиях, главным образом в скорлупе.
Токсическое действие меди является важным фактором, приводящим к развитию морфологических изменений в мозговой ткани.
Этиология и патогенез.
Заболевание наследуется по аутосомно-рецессивному типу.
Ген болезни расположен на длинном плече хромосомы 13, он клонирован и изучен. Ген кодирует переносящую медь-АТФазу, с которой связывается 6 атомов меди. К настоящему времени выявлено более 25 различных мутаций гена. Мутации на каждой из хромосом различны, что затрудняет установление соответствия между фенотипом и генотипом.
Существенно, что у гетерозиготных носителей заболевание не развивается.
Нормальная концентрация меди в плазме крови составляет 100—120 мкг%, причем 93% этого количества находится в виде церулоплазмина и только 7% связано с сывороточными альбуминами.
Медь в церулоплазмине связана прочно.
Связь осуществляется в печени.
Церулоплазмин является b2-глобулином, и каждая его молекула содержит 8 атомов меди.
Радиоизотопными методами установлено, что при гепатолентикулярной недостаточности нарушение связано с генетическим дефектом синтеза церулоплазмина, из-за чего содержание его резко снижено.
При этом медь не может быть стабильно связана и откладывается в тканях.
В некоторых случаях содержание церулоплазмина остается нормальным, но меняется его структура (изменяется отношение фракций). Особенно тропна медь при синдроме Вильсона — Коновалова к печени, ядрам мозга, почкам, эндокринным железам, роговице.
При этом медь начинает действовать как токсический агент, вызывая типичные дегенеративные изменения в этих органах.
Клиническая картина характеризуется разнообразием, что обусловлено повреждающим действием меди на многие ткани.
Преимущественное поражение того или иного органа зависит от возраста. У детей это в основном печень (печеночные формы).
В дальнейшем начинают превалировать неврологическая симптоматика и нарушения психики (нейропсихические формы).
Если заболевание проявляется после 20 лет, то у больного обычно отмечается неврологическая симптоматика.
Возможно сочетание симптомов обеих форм.
У большинства больных в возрасте от 5 до 30 лет уже выражены клинические проявления заболевания и диагноз установлен.
Кольцо Кайзера — Флейшера представляет собой зеленовато-коричневое кольцо по периферии роговицы.
Вначале оно появляется на верхнем полюсе.
Для выявления кольца Кайзера — Флейшера больной, как правило, должен быть осмотрен окулистом с помощью шелевой лампы.
Оно выявляется у больных, имеющих неврологические симптомы, и может отсутствовать у молодых больных с острым началом заболевания.
Течение. Различают острую и хроническую формы.
Острая форма характерна для раннего возраста, развивается молниеносно и кончается летально, несмотря на лечение.
Чаше встречается хроническая форма с медленным течением и постепенным развитием симптоматики.
Раньше всего появляется экстрапирамидальная мышечная ригидность нижних конечностей (нарушение походки и устойчивости).
Постепенно формируется картина паркинсонизма, затем изменяется психика (параноидальные реакции, истерия).
Иногда на первый план выступает печеночная недостаточность: увеличение печени, картина напоминает цирроз или хронический активный гепатит.
Диагностика. Ведущим признаком является гипокупремия ниже 10 мкг%, много меди выделяется с мочой — свыше 100 мкг/сут.
Может быть положительная тимоловая проба.
Важным симптомом является кольцо Кайзера — Флейшера.
Уровни церулоплазмина и меди в сыворотке обычно снижены, КТ черепа, выполненная еще до появления неврологических симптомов, может выявить увеличение желудочков, а также другие изменения.
МРТ обладает большей чувствительностью.
Она может выявить расширение III желудочка, очаги поражения в таламусе, скорлупе и бледном шаре.
Эти поражения обычно соответствуют клиническим проявлениям заболевания.
Генетические исследования (по Ш. Шерлок).
Братья и сестры больного должны быть обследованы.
О гомозиготности свидетельствуют гепатомегалия, спленомегалия, сосудистые звездочки, небольшое повышение активности трансаминаз в сыворотке.
Кольцо Кайзера — Флейшера выявляется не всегда.
Уровень церулоплазмина в сыворотке обычно снижен до 0,20 г/л и менее. Биопсия печени с определением содержания меди позволяет подтвердить диагноз.
Отличить гомозигот от гетерозигот легко, хотя иногда могут возникнуть трудности.
В таких случаях проводят анализ гаплотипов больного и его братьев и сестер.
Гомозигот лечат пеницилламином, даже если заболевание протекает бессимптомно.
Гетерозиготам лечение не требуется.
При наблюдении за 39 клинически здоровыми гомозиготами, получающими лечение, появления симптомов не отмечено, в то же время у нелеченых гомозигот развилась болезнь Вильсона и часть из них умерли.
Использование ДНК-маркеров позволяет с высокой точностью установить диагноз болезни Вильсона.
Однако генетическое исследование имеет значительные ограничения, в числе которых высокая стоимость методики и значительная вариабельность мутаций гена болезни Вильсона.
В настоящее время это исследование проводится у детей, чьи близкие родственники страдают заболеванием, и когда стандартные тесты не дают однозначного ответа, подтверждающего или отрицающего диагноз болезни Вильсона.
Патоморфология печени. Закономерно выявляется баллонная дистрофия, многоядерность гепатоцитов, скопления гликогена и гликогеновая вакуолизация ядер. Характерна жировая инфильтрация гепатоцитов. Клетки Купфера обычно увеличены в размерах.
У некоторых больных эти изменения особенно ярко выражены; выявляются тельца Мэллори, что напоминает морфологическую картину острого алкогольного гепатита.
У части больных наблюдаются изменения в печени, свойственные ХГ.
Гистологические изменения в печени при болезни Вильсона не являются диагностическими, однако выявление описанных выше изменений у молодых больных с циррозом печени позволяет заподозрить это заболевание.
Метод выявления меди окрашиванием рубеановой кислотой или родамином ненадежен, поскольку медь распределяется неравномерно и в узлах регенерации отсутствует.
Накопление меди обычно происходит в перипортальных гепатоцитах и сопровождается появлением атипичных отложений липофусцина.
Возможно определение содержания меди в печени.
Нормальное содержание меди в ткани печени 15—55 мкг на 1 г сухого вещества ткани печени.
У пациентов с болезнью Вильсона эти значения увеличены и колеблются от 250 до 3000 мкг/г.
Электронная микроскопия. Даже при бессимптомном течении заболевания выявляют аутофагические вакуоли и крупные измененные митохондрии. Жировая инфильтрация может быть связана с повреждением митохондрий. Можно видеть инфильтрацию межклеточного пространства волокнами коллагена, а также светлые и темные клетки печени.
Дифференциальную диагностику проводят с острым и хроническим гепатитом, при котором уровень церулоплазмина может быть снижен из-за нарушения его синтеза в печени.
Недоедание также способствует снижению уровня церулоплазмина.
При приеме эстрогенов, пероральных контрацептивов, при обструкции желчных путей, при беременности уровень церулоплазмина может повышаться.
Суточная экскреция меди при болезни Вильсона повышена.
Во избежание искажения результатов анализа рекомендуется собирать мочу в специальные бутылки с широким горлом с пакетами-вкладышами одноразового пользования, не содержащими медь.
При наличии противопоказаний к биопсии печени при нормальном уровне церулоплазмина в сыворотке заболевание можно диагностировать по степени включения в церулоплазмин перорально принимаемой радиоактивной меди.
Лечение. Требуется диета с исключением продуктов, содержащих большее количество меди (шоколад, какао, горох, печень, ржаной хлеб).
Лекарственная терапия проводится на протяжении всей жизни с момента устаноаления диагноза или обнаружения гомозиготного носительства дефектного гена и является залогом увеличения выживаемости. Необоснованное прекращение лечения может привести к необратимым изменениям и летальному исходу.
Препарат выбора — D-пеницилламин. До сих пор он остается средством выбора и «золотым стандартом» в лечении болезни Вильсона.
Механизмы действия D-пеницилламина: образование хелатных комплексов с медью, которые выделяются с мочой, и перевод внутриклеточной меди в неактивное состояние.
Препарат рекомендуется принимать натощак (за 30 мин до еды), так как пиша уменьшает его абсорбцию.
Учитывая, что D-пеницилламин дает антипиридоксиновый эффект, к терапии необходимо добавлять пиридоксин в дозе 25 мг/л внутрь.
Схема лечения:
Первый этап — начальная фаза лечения.
Начальная доза D-пеницилламина составляет 250—500 мг/сут, ее делят на 4 приема.
Затем дозу постепенно увеличивают до 1-2 г/сут (каждые 7 дней на 250 мг), пока экскреция меди с мочой не повысится до 2000—5000 мкг/сут.
После достижения клинического улучшения, которое наступает через несколько месяцев от начала лечения, и снижения экскреции меди с мочой переходят на поддерживающую терапию.
В течение двух первых месяцев лечения клинический анализ крови (количество форменных элементов) и мочи (величина протеинурии) проводят каждые 2 нед, в течение следующих 6 мес — ежемесячно.
Второй этап — поддерживающая терапия. Поддерживающие дозы составляют 0,75-1,25 г/сут. Экскреция меди с мочой уменьшается до 500— 1000 мкг/сут.
Ежегодно проводится исследование колец Кайзера — Флейшера в щелевой лампе.
При адекватном лечении происходит уменьшение выраженности и полное исчезновение симптома у 80% больных через 3—5 лет после начала лечения.
Побочные эффекты при лечении D-пеницилламином делятся на ранние, являющиеся в начальной фазе лечения, и поздние, развивающиеся во время поддерживающей терапии.
Ранние побочные эффекты. В течение первого месяца терапии у 20% больных наблюдается появление или ухудшение неврологической симптоматики. Это связано с мобилизацией меди из печени, повышением ее концентрации в ЦНС.
В этой ситуации необходимо снизить дозу до 250 мг/сут и постепенно повышать ее до увеличения экскреции меди с мочой.
Если неврологическая симптоматика продолжает ухудшаться, то D-пеницилламин заменяют другим медь-хелатирующим препаратом (см. ниже).
Ухудшение неврологической симптоматики в первые месяцы лечения необходимо дифференцировать с прогрессированием самого заболевания при применении низких доз D-пеницилламина.
В течение первого месяца лечения у 20% больных развиваются реакции гиперчувствительности — лихорадка, кожный зуд, сыпь и — редко — лимфаденопатия.
Эти симптомы проходят после временной отмены препарата.
Терапию D-пеницилламином возобновляют в дозе 250 мг/сут в комбинации с преднизолоном в дозе 20—30 мг/сут.
В течение месяца дозу D-пеницилламина увеличивают, постепенно отменяя преднизолон.
Поздние побочные эффекты. Развиваются у 5-7% пациентов и обычно манифестируют после года лечения. Наиболее частыми из них являются кожные изменения: пеницилламиновая дерматопатия, пемфигус, acantosis nigricans, elastosis perforans serpinginosa, lichen planus.
У 3-5% больных развиваются синдромы, сходные с аутоиммунными заболеваниями: синдромом Гудпасчера, системной красной волчанкой, миастенией.
При развитии этих осложнений, а также при появлении значительной протеинурии (более I г/сут) D-пеницилламин отменяют и назначают триентин.
Триентин. Используется с 1969 г. как альтернативный медьхелатирующий агент у пациентов, интолерантных к D-пеницилламину.
При переходе на триентин большинство побочных эффектов D-пеницилламина исчезает.
Дозы триентина составляют 1—2 г в день, разделенных на 3 приема. Препарат принимают натощак.
Наиболее тяжелым побочным эффектом является сидеробластная анемия.
Цинк. Использование цинка при болезни Вильсона основано на его способности увеличивать синтез медьсвязывающих белков в эпителии тонкой кишки и в гепатоцитах.
Это препятствует абсорбции меди из ЖКТ и обеспечивает перевод меди в нетоксичное состояние. Ежедневные дозы сульфата или ацетата цинка составляют 150 мг в день, разделенных на два-три приема.
Препарат назначают между приемами пищи.
Цинк относительно безопасен, из побочных эффектов наиболее частыми являются расстройства со стороны ЖКТ и головная боль.
Целесообразно использование цинка у асимптомных больных на ранних стадиях заболевания и в качестве поддерживающей терапии у пациентов, которым предварительно проводилась терапия медьхелатирующими препаратами.
Одновременное назначение хелаторов меди и препаратов цинка не рекомендуется.
Тетратиомолибдат. Механизмами действия этого препарата являются: образование комплексов с медью в ЖКТ и сыворотке крови, что препятствует соответственно ее абсорбции и проникновению в ткани. Рассматривается как потенциально более эффективный хелатор меди, чем D-пеницилламин и триентин.
В настоящее время имеются только ограниченные данные о клиническом использовании этого препарата.
Суточная доза составляет 120—200 мг. В качестве побочных эффектов описано угнетение костномозгового кроветворения.
Лечение хелаторами меди во время беременности не должно прекращаться. Рекомендуемые дозы D-пеницилламина, составляющие 0,75—1 г/сут, не представляют риска для плода.
Если планируется кесарево сечение, то за 6 нед до родоразрешения и на весь срок до заживания послеоперационной раны дозу D-пеницилламина необходимо снизить до 250 мг/сут.
Показаниями к трансплантации печени при болезни Вильсона являются: печеночная недостаточность, ассоциированная с гемолизом и гиперурикемней; прогрессирование печеночной недостаточности, не поддающейся медикаментозной коррекции.
Показатели однолетней выживаемости после трансплантации составляют около 80%.
источник