Заболевание, возникновение которого связано с изъянами в генетическом клеточном аппарате, — фенилкетонурия – входит в немногочисленный перечень наследственных болезней, поддающихся лечению. Первооткрывателем этого недуга был врач из Норвегии И.А. Феллинг, позже было выявлено, что за развитие и течение болезни отвечает единственный ген, именуемый геном фенилаланингидроксилазы (длинное плечо 12ой хромосомы, содержащей до 4,5% всего материала ДНК клетки). Наследуемый дефект приводит к частичной или полной деактивации фермента печени фенилаланин-4-гидроксилазы.
Наследственное заболевание фенилкетонурия (ФКУ) приводит к хроническому отравлению организма токсическими веществами, образованных вследствие нарушенного метаболизма аминокислот и процесса гидроксилирования фенилаланина. Постоянная интоксикация вызывает поражение центральной нервной системы (ЦНС), проявлением чего выступает прогрессирующее снижение интеллекта (фенилпировиноградная олигофрения).
Болезнь Феллинга проявляется в избыточном накоплении в организме фенилаланина и продуктов его неправильного обмена. К другим факторам развития фенилкетонурии относится нарушенное транспортирование аминокислот через гематоэнцефалический барьер, низкое количество нейротрансмиттеров (серотонин, гистамин, допамин). При отсутствии своевременного лечения заболевание приводит к умственной отсталости и может стать причиной гибели ребенка.
Причинообразующим фактором возникновения генных нарушений является метаболический блок, который препятствует образованию фенилаланин-4-гидроксилазы (фермент, отвечающий за превращение аминокислоты фенилаланина в тирозин). Протеиногенная аминокислота тирозин является составной частью белков и пигмента меланина, поэтому является необходимым элементом для функционирования всех систем организма, а ее нехватка приводит к ферментопатии.
Следствием подавления образования метаболита, вызванного мутационной инактивацией фермента, является активирование вспомогательных путей обмена фенилаланина. Ароматическая альфа-аминокислота в результате дефектных обменных процессов распадается на токсические производные, которые в нормальных условиях не образуются:
- фенилпировиноградную кислоту (фенилпируват) – жирно-ароматическая альфа-кетокислота, ее образование приводит к миелинизации отростков нейронов и слабоумию;
- фенилмолочную кислоту – продукт, образовавшийся при восстановлении фенилпировиноградной кислоты;
- фенилэтиламин – начальное соединение для биологически активных передатчиков электрохимических импульсов, повышает концентрацию дофамина, адреналина и норадреналина;
- ортофенилацетат – токсичное вещество, вызывающее нарушение обменных процессов жироподобных соединений в головном мозге.
Медицинская статистика свидетельствует о том, что патологически измененный ген присутствует у 2% населения, но при этом он никак не проявляет себя. Генетический дефект передается ребенку от родителей только при наличии заболевания у обоих партнеров, при этом младенец в 50 % случаев становится носителем мутировавшего гена, оставаясь здоровым. Вероятность того, что фенилкетонурия у новорожденных приведет к заболеванию, составляет 25%.
Болезнь Феллинга является генетическим отклонением, наследуемым по аутосомно-рецессивному типу. Такой тип наследования означает, что развитие признаков врожденного заболевания произойдет только при наследовании ребенком по одной дефектной генокопии от обоих родителей, которые являются гетерозиготными носителями видоизмененного гена.
Развитие врожденного заболевания в 99% случаев вызывает мутация гена, ответственного за кодирование фермента, обеспечивающего синтез фенилаланин-4-гидроксилазы (классическая фенилкетонурия). До 1% генетических заболеваний связаны с мутационными изменениями, происходящими в других генах, и вызывающих недостаточность дигидроптеридинредуктазы (ФКУ II типа) или тетрагидробиоптерина (ФКУ III типа).
Классическая форма генетического заболевания у детей в большинстве случаев проявляется во внешне различимых признаках, начиная с 3-9 месяца жизни. Новорожденные, имеющие дефектный ген, выглядят здоровыми, отличительной особенностью бывает специфический хабитус (внешний облик) ребенка. Выраженная симптоматика появляется через 6-12 месяцев после рождения.
ФКУ II типа характеризуется тем, что первые клинические симптомы появляются спустя 1,5 года с момента появления на свет. Признаки заболевания не исчезают после диагностирования генетических отклонений и начала диетотерапии. Этот вид врожденной болезни нередко приводит к летальному исходу на 2-3 году жизни ребенка. Самыми частыми симптомами ФКУ II типа являются:
- выраженные отклонения в умственном развитии;
- гиперрефлексия;
- нарушение двигательных функций всех конечностей;
- синдром бесконтрольных мышечных сокращений.
Клинические признаки мутационных изменений генов III типа сходны с заболеванием, протекающим по II типу. Дефицит тетрагидробиоптерина характеризуется триадой специфических симптомов:
- высокая степень умственной отсталости;
- явно уменьшенный размер черепа по отношению к другим частям тела;
- спастичность мускулатуры (при этом возможна полная обездвиженность конечностей).

В ходе клинических исследований и наблюдений были выдвинуты предположения, что влияние токсичных производных фенилаланинового обмена вызывает снижение интеллектуальных способностей, которое носит прогрессирующий характер и может привести к слабоумию (олигофрении, идиотии). Среди предполагаемых причин необратимых нарушений мозговой деятельности самой обоснованной считается вызванная снижением уровня тирозина нехватка нейромедиаторов, передающих импульсы между нейронами.
Точную причинно-следственную связь между наследственным заболеванием и нарушениями мозга до настоящего времени не выявлено, как и механизм развития вследствие фенилкетонурии таких психических состояний, как эхопраксия, эхолалия, приступов злости и раздражительности. Данные результатов анализов свидетельствую о том, что фенилаланин оказывает прямое токсическое действие на мозг, что тоже может вызывать снижение интеллекта.
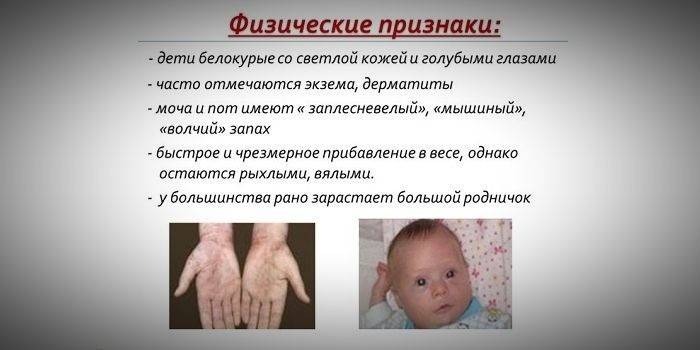
Ввиду того, что насыщенность пигментом кожи и волос зависит от уровня тирозина в митохондриях гепатоцитов, а фенилкетонурия приводит к остановке конверсии фенилаланина, пациенты с этим заболеванием имеют фенотипические особенности (рецессивные признаки). Повышенный мышечный тонус становится причиной появления отклонений в телосложении – оно становится диспластическое. К отличительным внешним признакам фенилкетонурии относятся:
- гипопигментация – светлая кожа, бледно-голубые глаза, обесцвеченные волосы;
- синюшность конечностей;
- уменьшенный размер головы;
- специфическое положение тела – при попытках стоять или сидеть ребенок принимает позу «портного» (руки и ноги согнуты в суставах).
При своевременном обнаружении болезнь Феллинга поддается успешному лечению путем корректировки питания, и развитие ребенка происходит в соответствии его возрастной группе. Трудность выявления генной мутации заключается в том, что ранние признаки тяжело обнаружить даже опытному педиатру. Выраженность симптоматики врожденного заболевания усиливается по мере взросления ребенка, потому что употребление белковой пищи способствует развитию нарушений ЦНС.
На протяжении первых дней жизни ребенка признаки патологических отклонений обнаружить трудно – малыш ведет себя естественно, задержки в развитии не наблюдается. Симптомы заболевания впервые начинают проявляться через 2-6 месяцев после рождения. Родителей должно насторожить поведение малыша, которое характеризуется низкой активностью, вялостью, или, наоборот, беспокойством, гипервозбудимостью.
С началом грудного вскармливания в организм новорожденного с молоком начинают поступать белки, что служит катализатором появления первых признаков, однозначно свидетельствующих о том, что заболевание начало прогрессировать. К специфическим клиническим проявлениям болезни относятся:
- постоянная рвота (зачастую принимаемая за врожденное сужение привратника);
- частое срыгивание;
- отсутствие реакции на внешние раздражители;
- мышечная дистония (сниженное напряжение мышц);
- судорожный синдром (судороги эпилептического или неэпилептического характера).
Если манифестация генетической болезни не произошла (или не была замечена) в течение первых 6 месяцев с момента рождения ребенка, то после этого периода уже можно точно определить отставание в психомоторном развитии. Симптомами генетических нарушений, вызванных ферментным дефицитом, у детей, старше полугода, являются:
- снижение активности (вплоть до полной безучастности);
- отсутствие попыток к самостоятельному вставанию, сидению;
- особенный «мышиный» запах кожи (запах плесени возникает вследствие выведения токсических производных фенилаланина через потовые железы и мочу);
- потеря способности к визуальному распознаванию лиц родителей;
- шелушение кожи;
- появление дерматитов, экзем, склеродермии.
Если отклонения в развитии не были выявлены в младенческом возрасте, и соответствующее лечение не проводилось, то заболевание начинает активно прогрессировать и нередко приводит к инвалидности. Отсутствие терапии на раннем этапе болезни вызывает появление следующих симптомов болезни уже в возрасте 1,5 лет:
- микроцефалия (уменьшенные размеры головного мозга);
- прогнатия (смещение верхнего зубного ряда вперед);
- позднее прорезывание зубов;
- гипоплазия эмали (истончение или полное отсутствие зубной эмали);
- задержка речевого развития вплоть до полного отсутствия речи;
- 3, 4 степень олигофрении (задержка психического развития, умственная отсталость);
- врожденные пороки сердца (дефекты в структуре сердечной мышцы, отделах сердца, крупных сосудах);
- расстройства вегетативной системы (акроцианоз, повышенная потливость, артериальная гипотония);
- запоры.
Для проявления мутации с аутосомно-рецессивным характером наследования дефектный ген должен быть унаследован от обоих родителей. Генетические заболевания такого типа встречаются с одинаковой частотой у новорожденных мальчиков и девочек. Патогенез ФКУ предопределяется нарушением обмена фенилаланина, который может протекать в 3 формах. Лечению диетотерапией поддается только классическая фенилкетонурия I типа.
Атипичные формы заболевания нельзя вылечить путем корректировки питания. Эти отклонения вызваны дефицитом тетрагидроптерина, дегидроптеринредуктазы (реже — пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и др.). Большинство случаев летальных исходов зафиксировано среди больных редкими вариациями ФКУ, при этом клинические проявления всех форм болезни аналогичны. Риск рождения ребенка с мутировавшим геном фенилаланингидроксилазы возрастает, если его родители являются близкими родственниками (при близкородственных браках).
При подозрении на генетические нарушения диагноз устанавливается на основании совокупности данных, полученных в результате изучения анамнеза болезни – генеалогических сведениях, результатах клинических и медико-генетических исследований. Для своевременного выявления врожденных заболеваний (ФКУ, муковисцидоза, галактоземии и др.) разработана программа обязательного массового обследования в лабораторных условиях всех новорожденных детей (неонатальный скрининг).
Если будущие родители знают о носительстве мутировавшего гена, современная медицина предлагает способы обнаружения дефекта на этапе беременности (дородовая диагностика плода инвазивным методом). Для разделения фенилкетонурии на виды по степени тяжести применяется условная классификация, которая основана на уровне фенилаланина в безфибриногенной жидкости, полученной из плазмы крови:
- Тяжелая фенилкетонурия – 1200 мкмоль/л.
- Средняя – 60-1200 мкмоль/л.
- Легкая (не требует лечения) – 480 мкмоль/л.
Выявление генетических отклонений происходит в несколько этапов. На первом этапе в роддоме у всех младенцев на 3-5 день жизни осуществляется забор периферической крови (из пятки) для исследований. Материал наносится на бумажный бланк и отправляется в биохимическую лабораторию, где происходит его биохимический анализ. На втором этапе скриннинг-теста определяется соответствие концентрации фенилаланина нормальному значению.
Если патологических изменений не обнаружено, диагностика завершается, о чем делается запись в карте ребенка. При наличии отклонений от нормы результаты диагностики направляются врачу-педиатру для обеспечения уточняющего исследования образца крови новорожденного. Здоровье малыша зависит от своевременного и точного выполнения всех мероприятий по выявлению отклонений. Если диагноз подтвердится после повторного скрининг-теста, родители ребенка будут направлены в клинику к детскому генетику для назначения лечения.
Повторная диагностика при обнаружении во время проведения первичного скрининг-теста отклонений от нормы осуществляется путем повторной сдачи анализов. Помимо определения содержания фенилаланина в крови к методам диагностики ФКУ у детей и взрослых относятся:
- проба Феллинга – определение фенилпировиноградной кислоты в моче путем добавления к биоматериалу хлорида железа (происходит окрашивание в сине-зеленый цвет);
- тест Гатри – оценка степени реакции микроорганизмов на продукты обмена или ферменты, содержащиеся в крови пациента;
- хроматография – изучение химических свойств веществ, распределенных между двумя фазами;
- флуориметрия – облучение биоматериала монохроматическим излучением для определения концентрации содержащихся в нем веществ;
- электроэнцефалография – диагностика электрической активности головного мозга;
- магнитно-резонансная томография – возбуждение атомных ядер клеток электромагнитными волнами и измерение их отклика.
В основе терапии фенилкетонурии лежит ограничение потребления продуктов, являющихся источником белков животного и растительного происхождения. Единственным способом успешного лечения является диетотерапия, адекватность которой оценивается по содержанию фенилаланина в сыворотке крови. Предельно допустимый уровень аминокислоты у больных разных возрастных групп составляет:
- у новорожденных и детей до 3 лет – до 242 мкмоль/л;
- у дошкольников – до 360 мкмоль/л;
- у пациентов в возрасте от 7 до 14 лет – до 480 мкмоль/л;
- у подростков – до 600 мкмоль/л.
От того, на каком этапе заболевания произведена коррекция рациона, зависит эффективность диеты. При ранней диагностике врожденной патологии диетотерапия назначается с 8 недели жизни (после этого периода уже начинаются необратимые изменения). Отсутствие своевременных мер приводит к осложнениям и снижению уровня интеллекта на 4 бала за 1 месяц с момента рождения до начала лечения.
Ввиду того, что лечебная диета при фенилкетонурии предполагает полное исключение из рациона животного белка, появляется необходимость применения других источников необходимых аминокислот, а также витаминов группы В, кальций- и фосфорсодержащих минеральных соединений. К продуктам, назначаемым в качестве добавок к безбелковому рациону, относятся:
- белковые гидролизаты (Амиген, Аминазол, Фибриносол);
- не содержащие фенилаланин смеси, насыщенные незаменимыми аминокислотами – Тетрафен, Фенил-фри.
Наряду с лечебными мерами по устранению причины нарушений функционирования организма следует проводить симптоматическое лечение, направленное на устранение дефектов речи и нормализацию координации движений. Комплексная терапия включает физиотерапевтические процедуры, массаж, помощь логопеда, психолога, выполнение гимнастических упражнений. В ряде случае совместно с диетотерапией показан прием антиконвульсантов, ноотропных и сосудистых препаратов.
Фенилкетонурия II и III типа не поддается лечению низкобелковой диетой – уровень фенилаланина в крови остается неизменным при ограничении поступления белка в организм или клиническая симптоматика прогрессирует даже при снижении уровня аминокислоты. Эффективная терапия этих форм заболевания осуществляется с применением:
- тетрагидробиоптерина – фактора пораженного фермента;
- синтетических аналогов тетрагидробиоптерина – эти вещества лучше проникают через гематоэнцефалический барьер;
- препаратов заместительной терапии – не устраняют причину фенилкетонурии, но поддерживают нормальное функционирование организма (Леводопа совместно с Карбидофой, 5-окситриптофан, 5-формилтетрагидрофолат);
- гепатопротекторов – поддерживают функционирование печени;
- противосудорожных средств;
- введения гена фенилаланингидроксилазы в печень – экспериментальный метод.
На первом году жизни ребенка с ФКУ допустимо кормление грудным молоком, но его количество должно быть ограничено. До 6 месяцев допустимым уровнем потребления фенилаланина является 60-90 мг на 1 кг веса малыша (в 100 г молока содержится 5,6 мг фенилаланина). Начиная с 3 месяцев, рацион ребенка следует поэтапно расширять, вводя в него фруктовые соки и пюре.
Детям с 6 месяцев разрешено введение в рацион овощных пюре, каш (из саго), безбелковых киселей. После 7 месяцев можно давать малышу низкобелковые макаронные изделия, с 8 месяцев – хлеб, не содержащий белка. Возраст, до которого следует ограничивать поступление белка в организм больного ребенка, не установлен. Врачи до настоящего времени дискутируют по вопросу целесообразности пожизненной диетотерапии, но сходятся во мнении, что минимум до 18 лет необходимо придерживаться диетического питания.
Фенилкетонурия, диагностированная у женщины, не является поводом отказаться от рождения ребенка. Будущим мамам с ФКУ для предупреждения повреждения плода во время беременности и профилактики возможных осложнений необходимо до планируемого зачатия и во время вынашивания ребенка придерживаться диеты с ограничением фенилаланина (его уровень в крови должен быть до 242 мкмоль/л).
Диета при фенилкетонурии базируется на существенном уменьшении дозы натурального белка в суточном рационе, но организм новорожденного ребенка не может развиваться нормально при отсутствии необходимых микроэлементов. Для восполнения потребности малыша в белке применяются безлактозные аминокислотные смеси, которыми, согласно российскому законодательству, больные должны обеспечиваться бесплатно.
Толерантность грудничка к фенилаланину в течение первого года жизни стремительно изменяется, поэтому необходимо контролировать его концентрацию в крови ребенка и вносить корректировки в диету. Смеси рассчитаны на определенные возрастные группы:
- малышам до года назначаются Афенилак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- детям, старше 1 года, назначают обогащенные витаминами и минералами смеси с повышенным содержанием белка – PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Одним из основных компонентов пищевой диеты при фенилкетонурии являются малобелковые продукты на основе крахмала. Эти добавки содержат гидролизат казеина, триптофан, тирозин, метионин, азот и обеспечивают суточную потребность организма ребенка в белке, который необходим для нормального развития и роста. Специализированными продуктами, восполняющими нехватку необходимых минералов и аминокислот при их нехватке в рационе питания, являются:
По мере адаптации организма к фенилаланину детям с возраста 5 лет можно постепенно уменьшать ограничения в питании. Расширение рациона происходит путем введения круп, молочных продуктов, мясных изделий. Школьники старших классов имеют уже высокую толерантность к фенилаланину, поэтому в этом возрасте можно продолжить расширение диеты, при этом необходимо отслеживать реакцию на все изменения в питании. Для контроля состояния ребенка применяются следующие способы:
- оценка неврологических показателей, психологического состояния;
- контроль показателей электроэнцефалограммы;
- определение уровня фенилаланина.
В рацион питания пациентов с ФКУ наряду с малобелковыми крахмалистыми продуктами и лечебными смесями входят и продукты натурального происхождения. При составлении меню следует четко рассчитывать количество потребляемого белка и не превышать рекомендованную врачом дозировку. Для исключения токсического влияния на организм разработаны 3 списка продуктов, которые содержат запрещенные (красный), нерекомендованные (оранжевый) и разрешенные (зеленый) позиции.
Фенилкетонурия развивается на фоне отсутствия фермента, превращающего фенилаланин в тирозин, поэтому высокое содержание белка является поводом для отнесения продуктов в запрещенный (красный) список. Позиции из этого перечня следует полностью исключить рациона питания больного ФКУ:
- мясо;
- внутренние органы животных, субпродукты;
- колбасы, сосиски;
- морепродукты (в т.ч. рыба);
- яйца всех птиц;
- кисломолочные продукты;
- орехи;
- плоды бобовых и зерновых культур;
- соевые продукты;
- желатинсодержащие блюда;
- кондитерские изделия;
- аспартам.
Продукты, которые должны дозировано поступать в организм ребенка с диагнозом ФКУ, включены в оранжевый список. Включение в рацион питания позиций из этого перечня допустимо, но в строго ограниченном количестве. Эти продукты хоть и содержат не много белка, но тоже могу повысить уровень фенилаланина, поэтому их употребление не рекомендовано:
- консервированные овощи;
- блюда из картофеля и риса;
- капуста;
- молоко;
- щербет.
Безбелковые продукты разрешены к употреблению больными с диагнозом фенилкетонурия без ограничений. Перед покупкой позиций из зеленого списка необходимо изучить состав, указанный на упаковке, и убедиться, что там не содержится краситель аспартам, содержащий фенилаланин:
- фрукты;
- овощи (за исключением картофеля и капусты);
- ягоды;
- зелень;
- крахмалистые крупы (саго);
- мед, сахар, варенье;
- мучные изделия из кукурузной или рисовой муки;
- масла, жиры (сливочное, растительное, оливковое).
Фенилкетонурия является неизлечимым заболеванием, которое можно перевести в фазу стагнации путем применения диетотерапии и лечебно-профилактических мер. При изменении условий жизни, нарушении режима питания болезнь может опять обостриться, поэтому больным требуется пожизненное наблюдение. Процесс контроля заключается в периодическом определении уровня фенилаланина в крови. Частота сдачи анализов зависит от возраста пациента:
- до 3 месяцев – скрининг крови надо делать еженедельно до получения устойчивых результатов;
- от 3 месяцев до 1 года – 1-2 раза в месяц;
- от 1 до 3 лет – 1 раз за 2 месяца;
- старше 3 лет – ежеквартально.

Кровь для анализов сдается через 3-4 часа после приема пищи. Помимо скрининга развитие ФКУ контролируется путем определения нутритивного статуса, физического, эмоционального развития больного, уровня интеллектуальных способностей и развитости речи. По результатам наблюдений может возникнуть необходимость дополнительной диагностики с привлечением соответствующих специалистов.
источник
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Фенилкетонурия — это клинический синдром, включающий умственную отсталость с когнитивными и поведенческими нарушениями, которые вызываются повышением уровня фенилаланина в крови. Первичной причиной является недостаточная активность фенилаланингидроксилазы. Диагноз основывается на выявлении высокого уровня фенилаланина и нормального или низкого уровня тирозина. Лечение состоит из пожизненного соблюдения диеты с низким содержанием фенилаланина. Прогноз отличный при своевременной диагностике.
 [1], [2], [3], [4], [5], [6], [7], [8], [9]
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Фенилкетонурия наиболее распространена среди белого населения и относительно реже встречается среди евреев ашкенази, китайцев и чернокожих. Тип наследования аутосомно-рецессивный; частота составляет примерно 1/10 000 рождений среди белого населения.
Избыток фенилаланина, поступающий с пищей (т. е. который не используется для синтеза белка), в норме превращается в тирозин под действием фенилаланингидроксилазы; тетрагидробиоптерин (ВН4) является необходимым кофактором для этой реакции. Если в результате одной или нескольких мутаций развивается дефицит или отсутствие фенилаланингидроксилазы, происходит накопление фенилаланина, поступающего с пищей; главным органом, страдающим от повышенного уровня фенилаланина, является головной мозг, вследствие нарушения миелинизации. Часть избыточного фенилаланина превращается в фенилкетоны, которые выводятся с мочой, что и обусловило название фенилкетонурия. Степень дефицита фермента и следовательно тяжести гиперфенилаланинемии варьирует среди пациентов в зависимости от конкретной мутации.
Большая часть детей рождается нормальными, однако у них в течение нескольких месяцев медленно развиваются симптомы фенилкетонурии, что связано с постепенным накоплением фенилаланина. Отличительным признаком фенилкетонурии при отсутствии лечения является выраженная задержка умственного развития. У детей также отмечаются выраженная гиперактивность, нарушение походки и психозы, а также неприятный, мышиный запах тела, появляющийся в результате выделения с мочой и потом фенилуксусной кислоты (продукт распада фенилаланина). У пациентов также отмечается тенденция иметь более светлые кожу, волосы и глаза, чем у здоровых членов семьи, у некоторых может появляться сыпь, похожая на инфантильную экзему.
Несмотря на то что практически все случаи (98-99 %) фенилкетонурии являются следствием дефицита фенилаланингидроксилазы, также фенилаланин может накапливаться, если не синтезируется ВН4 вследствие дефицита дегидробиптеринсинтетазы или не восстанавливается в связи с дефицитом дигидроптеридинредуктазы. В дополнение, поскольку ВН4 также является кофактором для тирозингидроксилазы, которая участвует в синтезе допамина и серотонина, дефицит ВН4 нарушает синтез нейротрансмиттеров, вызывая неврологические симптомы, вне зависимости от накопления фенилаланина.
[10], [11], [12], [13]
В США и многих развитых странах все новорожденные проходят скрининговое обследование на фенилкетонурию через 24-48 часов после рождения с применением одного из нескольких методов скрининга в крови; при получении результатов, отличающихся от нормальных, диагноз подтверждают непосредственным измерением уровня фенилаланина. При классической фенилкетонурии у пациентов часто уровень фенилаланина превышает 20 мг/дл (1,2 мкмоль/л). При частичном дефиците уровень фенилаланина обычно составляет менее 8-10 мг/ дл, если ребенок получает обычную пищу (уровень более 6 мг/дл указывает на необходимость лечения); дифференциальный диагноз с классической фенилкетонурии требует определения активности печеночной фенилаланингидроксилазы, при котором ее уровень оказывается равным 5-15 % от нормального, или мутационного анализа, идентифицирующего легкие мутации гена. Дефицит ВН4 отличается от других форм фенилкетонурии повышением концентрации биоптерина или неоптерина в моче, крови или ликворе или во всех этих биологических жидкостях; выявление этой формы важно, так как стандартное лечение фенилкетонурии не предотвращает повреждения головного мозга в этих случаях.
У детей из семей с положительным семейным анамнезом фенилкетонурия может выявляться пренатально при использовании прямого исследования мутаций после хорионбиопсии или амниоцентеза.
[14], [15], [16], [17], [18], [19], [20], [21], [22]
Лечение состоит из пожизненного ограничения поступления с пищей фенилаланина. Все естественные белки содержат около 4 % фенилаланина, поэтому диета должна включать низкобелковые продукты (например, фрукты, овощи, определенные злаки); белковый гидролизат, обработанный для того, чтобы удалить фенилаланин; и смеси аминокислот, которые не содержат фенилаланин. Примерами коммерческих продуктов, не содержащих фенилаланина, являются XPhe продукты (ХР Analog для грудных детей, ХР Maxamaid для детей от 1 года до 8 лет, ХР Maxamum для детей старше 8 лет); Phenex I и II; PhenilFree I и II; ФКУ1, 2, 3; PhenylAde (различные); Loflex; PlexylO. Небольшое количество фенилаланина необходимо для роста и обмена веществ; оно вводится дополнительно мерными порциями естественного белка в виде молока или низкобелковых продуктов.
Постоянный контроль за уровнем фенилаланина в крови необходим; рекомендуемый уровень составляет 2-4 мг/дл (120-240 мкмоль/л) для детей младше 12 лет и 2-10 мг/дл (120-600 мкмоль/л) для детей старше 12 лет. Планирование питания и лечение необходимо начинать у женщин детородного возраста до наступления беременности для того, чтобы обеспечить хороший исход для ребенка.
У детей с дефицитом ВН4 лечение также включает тетрагидробиоптерин 1-5 мг/кг внутрь 3 раза в день; леводопу, карбидопу и 5ОН триптофан и фолиевую кислоту 10-20 мг внутрь один раз в день в случаях дефицита дигидроптеридинредуктазы. Тем не менее цели и подходы к лечению такие же, как при фенилкетонурии.
Если адекватное лечение начинается в первые дни жизни ребенка, фенилкетонурия не развивается. Лечение фенилкетонурии, начатое после 2-3 лет, может быть эффективным только для контроля выраженной гиперактивности и некупируемого судорожного синдрома. Дети, рожденные матерями с плохо контролируемой фенилкетонурии (т.е. с наличием высокого уровня фенилаланина) во время беременности, имеют высокий риск развития микроцефалии и задержки развития.
источник
Фенилкетонурия (ФКУ) — это наиболее распространенное врожденное нарушение метаболизма аминокислот, возникающее в результате дефицита фермента фенилаланингидроксилазы. Вследствие данного заболевания снижается способность организма к метаболизму аминокислоты фенилаланина. Эти процессы вызывают накопление фенилаланина в тканях организма и физиологических жидкостях.
Повышенные уровни фенилаланина негативно влияют на когнитивные функции, поэтому у людей с классической фенилкетонурией почти всегда присутствует интеллектуальная инвалидность. Заболевание доходит до этой стадии, если уровни фенилаланина не контролируются путем диетической или медикаментозной терапии.
Механизм, вследствие которого повышенные уровни фенилаланина вызывают интеллектуальную инвалидность, в точности не известен, однако ограничение продуктов, содержащих вредные вещества, снижает этот эффект.
В детском возрасте уровень фенилаланина в крови и коэффициент интеллекта (IQ) тесно связаны.
В настоящее время в ведущих клиниках мира ведется детальное изучение незначительных нейропсихологических дефицитов у детей с контролируемой фенилкетонурией. Некоторые ученые списывают этот дефицит на остаточные аномалии функции нейромедиаторов — например, снижение производства нейротрансмиттеров в результате недостаточного транспорта тирозина сквозь нейронную клеточную мембрану.
Иногда уровни аминокислот у больных в норме, но присутствует дефицит синтеза или утилизации белка BH4. Такое состояние называется злокачественной фенилкетонурией. BH4 кофактор необходим для гидроксилирования тирозина (предшественника дофамина) и триптофана (предшественника серотонина). Таким образом, пациенты с дефицитом BH4 могут иметь дополнительные проблемы неврологического характера, которые невозможно полностью скорректировать диетическим ограничением количества фенилаланина. Таким пациентам необходимы дополнительные методы лечения, которые не гарантируют стопроцентной эффективности.
Фенилкетонурия — это аутосомно-рецессивное заболевание, вызванное мутацией в гене ФАГ (фенилаланингидроксилазы). На сегодняшний день учеными идентифицировано более 500 различных мутаций гена ФАГ.
Для фенилкетонурии характерна заметная генотипическая гетерогенность, как внутри определенной популяции, так и между различными популяционными группами населения. Генотип и фенотип людей с данным заболеванием тесно связаны, кроме того, неродственные индивидуумы с одинаковыми мутациями имеют определенную степень изменчивости толерантности к фенилаланину. Генотип — это совокупность наследственных генетических характеристик, а фенотип — это совокупность внешних факторов среды, влияющая на человека уже после рождения.
ФКУ является наследственным заболеванием, вызванным дефектом в гене ФАГ. Ген ФАГ помогает создать фенилаланингидроксилазу — фермент, ответственный за разрушение фенилаланина. Опасное накопление фенилаланина может произойти, если человек употребляет чрезмерное количество продуктов с высоким содержанием белка, например, яйца и мясо. Оба родителя должны иметь дефектную версию гена ФАГ, только тогда ребенок наследует расстройство. Если только один родитель является носителем измененного гена, ребенок не будет иметь каких-либо симптомов, но так же будет являться носителем гена.
Скрининг-тест выполняется, когда новорожденному исполняется один-два дня, и он еще находится в роддоме.
Дополнительные тесты могут быть выполнены, чтобы подтвердить первоначальные результаты. Обычно врачи проверяют ген ФАГ на наличие мутаций, которые вызывают фенилкетонурию.
Если ребенок или взрослый проявляет симптомы фенилкетонурии, например, очевидна задержка развития, необходимы дополнительные анализы крови для подтверждения диагноза. Тест подразумевает взятие образца крови и ее анализ на присутствие фермента, необходимого для расщепления фенилаланина.
Большинство детей с фенилкетонурией кажутся нормальными при рождении. Если анализ крови после рождения не был проведен, а заболевание осталось невыявленным, происходит прогрессивная задержка развития — наиболее распространенный симптом фенилкетонурии.
Первые симптомы фенилкетонурии у новорожденных:
- рвота;
- судороги;
- неприятный запах от тела, так называемый «мышиный» запах (из-за выделения фенилуксусной кислоты);
- экзема;
- микроцефалия;
- расстройства поведения (раздражительность, плаксивость, отказ от груди). Ребенок плохо спит, его поведение отличается особым беспокойством. Такие дети по мере взросления становятся буквально неуправляемыми,
У пожилых людей с фенилкетонурией на МРТ отчетливо видны проявления демиелинизации. Присутствует заторможенность когнитивных функций и двигательных навыков. Коэффициент умственного развития снижается на 10 единиц и более, если прекратить придерживаться рекомендуемой врачом диеты.
- изменение цвета волос и оттенка кожи, являющееся следствием ухудшения синтеза меланина.
Больной имеет волосы светло-рыжего или белого цвета и болезненно бледную кожу, голубые глаза, светлые ресницы и брови.
Такое изменение цвета волос и кожи может быть характерным даже для афроамериканцев и азиатов, однако это скорее редкость, чем норма.
Другие симптомы кожных проявлений фенилкетонурии:
- экзема (атопический дерматит);
- увеличение числа гнойных инфекций;
- фолликулярный кератоз;
- потеря волос.
Симптомы фенилкетонурии могут быть как легкими, так и интенсивными. Наиболее тяжелой формой расстройства является так называемая классическая фенилкетонурия. Ребенок с классическим типом заболевания может казаться абсолютно нормальным первые несколько месяцев жизни. Если заболевание не выявляется и не лечится, появляются первые симптомы, которые затем усиливаются.
К таким симптомам относятся:
- аномалии функции глаз;
- паркинсонизм;
- гипопигментация кожи;
- «мышиный» запах, характерный для мочи и пота;
- интеллектуальная инвалидность;
- эпилепсия;
- тремор конечностей;
- задержка роста;
- гиперактивность;
- кожные заболевания;
- неприятный запах изо рта;
- неприятный запах изо рта;
Менее тяжелая форма фенилкетонурии — это состояние, при котором у новорожденного в организме слишком большое количество фенилаланина. Дети с этой формой заболевания могут иметь только легкие симптомы, но они должны придерживаться специальной диеты, чтобы избежать развития умственных нарушений.
Если фенилкетонурия не диагностируется при рождении и лечение не начинается быстро, расстройство может вызвать:
- необратимое повреждение мозга и психические расстройства в течение первых нескольких месяцев жизни;
- поведенческие проблемы и судороги у детей более старшего возраста.
Пациенты с фенилкетонурией могут облегчить тяжесть симптомов и предотвратить развитие осложнений, следуя специальной диете и принимая лекарства.
Самой эффективной диетой является ограничение продуктов, содержащих фенилаланин. Дети с ФКУ не могут потреблять грудное молоко, они питаются по специальной схеме, употребляя продукт Лофеналак или его аналоги — продукты, специально предназначенные для людей с таким заболеванием.
Если ребенок достаточно взрослый, чтобы жевать твердую пищу, необходимо полностью исключить такие продукты:
Для того чтобы больные могли получать достаточное количество белка, дети с ФКУ употребляют специальные пищевые добавки, содержащие все аминокислоты, необходимые организму, за исключением фенилаланина.
Существуют также определенные продукты с низким содержанием белка, которые можно найти в специализированных магазинах или аптеках. Эти пищевые добавки больные должны употреблять всю жизнь, чтобы избежать снижения умственных способностей и появления различных неприятных симптомов.
Все пациентам с фенилкетонурией необходимо постоянно контактировать с диетологом или лечащим врачом, чтобы правильно поддерживать надлежащий баланс питательных веществ, ограничивая при этом потребление фенилаланина. Также необходимо контролировать уровни фенилаланина путем ведения учета количества фенилаланина в продуктах питания, которые человек потребляет в течение дня.
Управление качеством пищевых продуктов и медикаментов США (FDA) недавно одобрило препарат Сапроптерин (Kuvan) для лечения фенилкетонурии. Sapropterin помогает снизить уровень фенилаланина. Этот препарат должен быть использован в сочетании со специальной схемой употребления пищи. Однако наиболее эффективен он для людей с легкими формами фенилкетонурии.
Беременность и фенилкетонурия
Женщина с фенилкетонурией в период беременность подвергается риску осложнений, если не следует схеме питания, рекомендованной врачом. Среди возможных рисков следует отметить вероятность выкидыша. Также есть шанс, что пока еще нерожденный ребенок будет подвергаться воздействию высоких уровней фенилаланина. Это может привести к различным проблемам у плода, в том числе:
- ограниченным интеллектуальным возможностям;
- порокам сердца;
- задержке роста;
- низкому весу на момент рождения;
- аномально маленькой голове (микроцефалии).
Все эти признаки становятся заметными у новорожденного не сразу, но тест, проведенный в первые сутки – двое после рождения, может помочь определить наличие заболевания.
Иногда лечение больных с фенилкетонурией (ФКУ) проводят в специальных клиниках, профилем которых являются метаболические нарушения. Обычно схема лечения разрабатывается комплексно, при участии эндокринолога, иммунолога и диетолога.
Уровни фенилаланина следует проверять через регулярные промежутки времени, примерно 1-2 раза в неделю у новорожденных и один раз в месяц для детей старшего возраста и взрослых.
Большинство американских клиник рекомендуют удерживать уровень фенилаланина в пределах 2-6 мг/дл (120-360 мкмоль/л). Точное попадание в рекомендуемый диапазон показателей требует квалифицированного ухода и тщательного мониторинга.
Диету ни в коем случае не рекомендуется прекращать по достижении подросткового возраста, поскольку гиперфенилаланинемия может иметь пагубные последствия и для взрослых пациентов.
Некоторые взрослые пациенты с незарегистрированной фенилкетонурией и, следовательно, интеллектуальной инвалидностью, могут значительно улучшить когнитивные показатели и физическое состояние при соблюдении специальной диеты.
Больные, проходящие качественное лечение, могут придерживаться нормального уровня активности.
Прогноз для разных групп пациентов сильно отличается. Например, дети, заболевание которых было обнаружено в раннем возрасте (в течение первого месяца жизни), при тщательном контроле состояния здоровья, соблюдении режима питания, могут жить абсолютно нормальной жизнью.
Если ребенок не придерживается диеты или соблюдает её частично, нарушения умственного развития неизбежны.
Большинство больных с фенилкетонурией, не имеющих никакого лечения, принадлежат к категории людей с глубокой интеллектуальной инвалидностью.
Распространенные трудности таких больных это:
- психологические проблемы, например, агорафобия и другие фобии;
- нарушение внимания;
- пониженная способность к концентрации;
- нарушения поведения.
Долгосрочный прогноз для пациентов с ФКУ очень хороший, если сразу после рождения ребенка родители придерживаются специальной диеты. Интеллектуальная инвалидность без лечения наступает уже в первый год жизни.
Проявляется она в следующем:
- заторможенность;
- нарушения поведения;
- неспособность к адекватному выражению эмоций (ребенок не узнает родителей, капризничает и т.д.);
- неврологические проблемы (судороги, тремор).
Можно ли предотвратить фенилкетонурию?
ФКУ является генетическим заболеванием, поэтому она не может быть предотвращена. Однако, ферментный анализ — это один из способов узнать, являются ли муж или жена носителем дефектного гена.
Всем больным с ФКУ следует избегать аспартама (искусственного подсластителя). Аспартам широко используется в производстве лекарственных средств, витаминов, напитков и других съедобных продуктов.
Диета с исключением фенилаланина увеличивает вероятность метаболических проблем, например, снижает способности организма к усвоению фолиевой кислоты или жирных кислот.
По материалам:
© 2005 — 2016 Healthline Media.
© 1998-2016 Mayo Foundation for Medical Education and Research.
Georgianne L Arnold, MD; Chief Editor: Luis O Rohena, MD.
В каких случаях опасно использовать масло и воду при ожогах?
источник
Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллинга) – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. В результате накопления токсических продуктов из-за неправильного метаболизма развивается отставание в умственном и физическом развитии.
Причем появившиеся нарушения в состоянии здоровья необратимы, но своевременная диагностика заболевания может предотвратить все патологические изменения. Лечение заключается в исключении продуктов питания, содержащих фенилаланин. Если такая элиминационная диета применяется практически с рождения, человек вырастает здоровым.
Давайте узнаем поподробнее, что же это за заболевание, чем оно проявляется, как диагностируется и лечится.
Впервые описана в 1934 г. доктором Феллингом, откуда и получила свое второе название. Встречается с частотой в среднем 1: 10 000, но в разных странах мира существуют колебания от 1:2600 (в Турции) до 1:100 000 (в Финляндии и Японии), в России от 1:5 000 до 1:10 000.

В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия.
Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени.
Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.
Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.).
Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III.
Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
 У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей.
Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами.
Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах.
Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
 Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики. Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина. Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.
Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу.
Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции).
Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека.
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ.
Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты.
Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально.

В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.

Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Если женщина, страдающая фенилкетонурией, при планировании беременности и при ее наступлении не придерживается соблюдения диеты, то это отражается на развитии ее ребенка. Потомство таких женщин имеет задержку внутриутробного развития и врожденные пороки развития: пороки сердца, аномалии развития головного мозга, мочевого пузыря, микроцефалию, аномалии лицевого скелета (расщелины).
Чтобы предотвратить патологические изменения у ребенка, таким женщинам необходимо придерживаться элиминационной диеты до зачатия и всю беременность. Дефицит белка восполняется за счет специальных белковых смесей без фенилаланина.
Таким образом, фенилкетонурия – это генетическое нарушение аминокислотного обмена, которое при поздней диагностике приводит к развитию выраженных интеллектуальных нарушений у ребенка.
Скрининг этого заболевания в роддомах позволяет диагностировать его в первые недели жизни и вовремя назначить лечение.
Основным методом в настоящее время является назначение элиминационной диеты, которая позволяет сохранить интеллект маленькому человечку, а значит, сохранить здоровье, что обеспечит полноценное существование в течение всей жизни.
Уважаемые родители!
Фенилаланин (ФА) — это эгзогенная незаменимая аминокислота, необходимая для нормального роста и развития, которая поступает в организм с пищей. У пациентов с ФКУ доза фенилаланина ограничивается до количества, которое зависит от индивидуальной толерантности к ФА.
Низкобелковая диета позволяет удерживать концентрацию ФА в сыворотке крови больного уровне на безопасном для ЦНС уровне. Этот уровень определен для каждой возрастной группы. Для грудного возраста ФА должен быть на уровне 2-4мг%, проверка уровня концентрации ФА в сыворотке крови пациента проводится 1 раз/в неделю до 6 месяца жизни.
Фенилаланин находится во всех пищевых продуктах, содержащих белок. Поэтому пища с высоким содержанием белка должна быть вычеркнута из рациона питания больных с фенилкетонурией. Однако невозможно полностью исключить поступление фенилаланина в организм в связи с его значительной ролью в процессе роста и развития.
Чтобы содержание фенилаланина находилось на определённом „безопасном” уровне, диета должна состоять из лечебных препаратов с низким содержанием фенилаланина или без него (которые удовлетворяют потребность в белке на 70-80%), и такого количества натуральных продуктов, чтобы удовлетворить потребности организма в белке, минеральных компонентах, витаминах и фенилаланине, учитывая основные возрастные потребности ребёнка. Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза. Диета при ФКУ — это:
- Уменьшение дозы фенилаланина согласно индивидуальной толерантности фенилаланина, что означает уменьшение дозы натурального белка в суточном рационе
- Обеспечение соответствующей для нормального развития дозы белка (дополнительный белок без фенилаланина) из продуктов лечебного питания ФКУ
- Обеспечение соответствующей дозы энергии с использованием специальных низкобелковых продуктов
- Обеспечение соответствующей дозы витамин, макро- и микроэлементов – главным образом из препаратов ФКУ и других источников.
Диета с ограниченным (пониженным) содержанием фенилаланина в значительной степени ограничивает возможность выбора натуральных продуктов питания:
|
|
|
У пациентов с ФКУ количество потребляемого белка из натуральных продуктов не может превысить установленной нормы. В связи с этим у маленьких детей и у старших преобладающая часть потребности в белке, т.е. около 80%, должно быть погашено смесями, не содержащими фенилаланин, обогащёнными минеральными ингредиентами.
 Диета грудного ребёнка с ФКУ базируется на смесях (препаратах) без фенилаланина, которые являются главными источниками белка, витаминов, микро- и макроэлементов. Грудное молоко и молочная смесь для младенцев дополняют эту диету необходимым для роста фенилаланином. Количество препарата ФКУ, грудного молока или молочных смесей надо систематически изменять в зависимости от индивидуальной толерантности фенилаланина, а также и от потребностей растущего организма. В течение первого года жизни толерантность фенилаланина быстро изменяется, постоянно уменьшаясь, в связи с этим концентрацию фенилаланина в крови необходимо контролировать в определённых промежутках времени, а диету модифицировать.
Диета грудного ребёнка с ФКУ базируется на смесях (препаратах) без фенилаланина, которые являются главными источниками белка, витаминов, микро- и макроэлементов. Грудное молоко и молочная смесь для младенцев дополняют эту диету необходимым для роста фенилаланином. Количество препарата ФКУ, грудного молока или молочных смесей надо систематически изменять в зависимости от индивидуальной толерантности фенилаланина, а также и от потребностей растущего организма. В течение первого года жизни толерантность фенилаланина быстро изменяется, постоянно уменьшаясь, в связи с этим концентрацию фенилаланина в крови необходимо контролировать в определённых промежутках времени, а диету модифицировать.
 Начиная со второго года жизни, лечебную аминокислотную смесь ФКУ без фенилаланина постепенно сменяют смесями без фенилаланина с увеличенным содержанием белка. Состав этих смесей разработан с учетом потребностей в основных питательных веществах здоровых детей в соответствующих возрастных группах, за исключением фенилаланина: только смесь аминокислот («белковый эквивалент») в сочетании с витаминами и минералами. Необходимое количество белка свободного от фенилаланина поступает в организм из низкоэнергетических напитков: фруктовые соки, фруктово-овощные соки. Такая смена режима и рациона питания может повлиять на обмен аминокислот. Оптимальное использование свободных аминокислот из смеси для синтеза собственного белка возможно лишь при упртреблении соответствующего количества калорий за один приём в суточном рационе, ибо каждый процесс синтеза в организме человека осуществляется с использованием энергии.
Начиная со второго года жизни, лечебную аминокислотную смесь ФКУ без фенилаланина постепенно сменяют смесями без фенилаланина с увеличенным содержанием белка. Состав этих смесей разработан с учетом потребностей в основных питательных веществах здоровых детей в соответствующих возрастных группах, за исключением фенилаланина: только смесь аминокислот («белковый эквивалент») в сочетании с витаминами и минералами. Необходимое количество белка свободного от фенилаланина поступает в организм из низкоэнергетических напитков: фруктовые соки, фруктово-овощные соки. Такая смена режима и рациона питания может повлиять на обмен аминокислот. Оптимальное использование свободных аминокислот из смеси для синтеза собственного белка возможно лишь при упртреблении соответствующего количества калорий за один приём в суточном рационе, ибо каждый процесс синтеза в организме человека осуществляется с использованием энергии.
 В связи с тем, что смеси свободные от фенилаланина содержат мало калорий, а питание должно быть сбалансированным, т.е. отвечать определённым соотношениям содержания жиров и углеводов (основных источников энергии) таким образом, чтобы полностью удовлетворить суточную потребность организма в энергии, целесообразным становится обогащение суточного рациона иными высокоэнергетическими пищевыми прдуктами. Это стало возможным благодаря наличию на рынке большого количества специализированных низкобелковых и частично свободных от фенилаланина продуктов.
В связи с тем, что смеси свободные от фенилаланина содержат мало калорий, а питание должно быть сбалансированным, т.е. отвечать определённым соотношениям содержания жиров и углеводов (основных источников энергии) таким образом, чтобы полностью удовлетворить суточную потребность организма в энергии, целесообразным становится обогащение суточного рациона иными высокоэнергетическими пищевыми прдуктами. Это стало возможным благодаря наличию на рынке большого количества специализированных низкобелковых и частично свободных от фенилаланина продуктов.

Дневная доза смеси ФКУ зависит от возраста ребёнка, массы тела, общего состояния здоровья и индивидуальной суточной толерантности фенилаланина. Очень важно, чтобы рекомендуемое суточное количество смеси не давать в один приём, напр.
утром. Такой способ подачи смеси может привести к колебаниям аминокислотного равновесия или к симптомам нетолерантности препарата. Суточную дозу смеси необходимо поделить на 3-4 приёма в течении дня. Препарат следует принимать во время еды.
Суточное потребление фенилаланина из пищевых продуктов должно быть ограничено до такого количества, чтобы контролируемый уровень концентрации фенилаланина в сыворотке крови не превышал „безопасного для ЦНС” уровня, т.е.
2-4мг/дл, это и есть индивидуальная суточная толерантность фенилаланина.
С целью полного удовлетворения потребностей ребёнка с фенилкетонурией, и поддержания на допустимом уровне употребляемого из продуктов натурального белка и фенилаланина следует все пищевые продукты отмерять и взвешивать, а также выбирать продукты с самым низким содержанием фенилаланина.
Учитывая факт, что диета пациентов с ФКУ должна предусматривать ограничение дозы фенилаланина до количества безопасного для ЦНС, очень важно одновременно обеспечить поставку основных ингредиентов питания: белка, углеводов, жиров, а также витаминов, микро- и макроэлементов, энергии и объема жидкости согласно рекомендациям для определенных возрастных групп.
Очень часто повышение концентрации фенилаланина в сыворотке крови ребёнка означает, что количество употребляемого ребёнком фенилаланина значительно превышает рекомендуемую суточную дозу. Причиной может быть также проблема с употреблением препарата ФКУ.
Хронический недостаток энергии, также как и недобор белка ускоряют процесс разрушения собственных белков организма (катаболические процессы).
Разрушение белков и увеличение концентрации фенилаланина могут быть вызваны инфекционным процессом, который протекает с повышением температуры тела, рвотой, поносом, снижением аппетита, хирургическими операциями (интенсификация катаболических процессов, увеличение потребности в энергии).
В таких случаях следует увеличить поступающее в организм количество энергии. Во время болезни ребёнка следует обратить внимание на количество употребляемых калорий, так как дефицит энергии является основной причиной ускорения катаболизма белков, а вследствие этого повышается уровень фенилаланина.
При инфекциях следует увеличить поступление энергии на 20 – 30%. При высокой температуре необходимо увеличить количество энергии на 12% на каждый 1 градус температуры. При поносе или рвоте следует на 1-2 дня отказаться от диеты ФКУ, а после выздоровления постепенно к ней вернуться.
При быстром приросте массы тела ребёнка может появиться необходимость в дополнительной порции питания или препарата. Невнимательный подход к этим потребностям и несоблюдение основных требований диеты может отрицательным образом повлиять на уровень фенилаланина в крови пациента.
Родители должны постоянно восполнять свои знания о ФКУ и использовать их в практике ежедневной диеты. Обучая ребёнка правилам правильного питания, родители должны подчёркивать значимость систематического приёма пищи и употребления исключительно разрешенных продуктов. Ребенку нужно систематически объяснять в доступной форме необходимость отказа от высокобелковых продуктов. Важно также, чтобы ребёнок мог отказаться от угощения ровесников и умел определять сходства и отличия в выборе продуктов.

«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше…
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты.
Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ.
В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин.
В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта.
Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно.
Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* | Уровень фенилаланина в крови, мкмоль/л | Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) | 120–600 | 2–10 |
| Умеренная (мягкая, средняя) | 600–1200 | 10–20 |
| Классическая (тяжелая) | >1200 | >20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название | Причинный фермент |
| ФАГ*-зависимая ФКУ** | Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) | 6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B | Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) | Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D | Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит | Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях.
У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина.
Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести.
Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов).
Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина. Также на количество медиаторов ЦНС влияет и само количество фенилаланина.
Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.

Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным».
Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии.
Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии.
При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин.
Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
- При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
- Лечение
- Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка =
50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму. Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока.
- Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] — Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] — М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend
Все формы ФКУ можно диагностировать уже в первые недели или даже дни жизни ребенка, когда клинические проявления еще отсутствуют. Для этого проводят биохимический скрининг новорожденных на наличие гиперфенилаланинемии.
В настоящее время, согласно приказу Минздрава России № 316 от 30.12.93 проведение неонатального скрининга на ФКУ стало обязательным.
В практике массового обследования новорожденных на фенилкетонурию используются разные методические подходы:
1. Тест Гатри – полуколичественный микробиологический тест для
определения концентрации фенилаланина в крови. В его основе лежит ингибирование бактериальной культуры Bacillus subtilis.
Тест Гатри до настоящего времени используется во многих странах для проведения скрининга новорожденных. По данным W.B.Hanley (1997) чувствительность теста Гатри составляет 99,2%.
В РФ метод не применяется.
2. Хроматография – полуколичественный биохимический метод определения фенилаланина с помощью тонкослойной хроматографии аминокислот плазмы крови и мочи.
В РФ в качестве массового скрининга метод не применяется.
3. Флюориметрия – количественный биохимический метод определения фенилаланина в крови методом хроматографии с помощью современных автоматических флюориметров.
Используется во многих развитых странах (в том числе и России) для проведения массового автоматизированного скрининга.
4. Тандемная масс-спектрометрия – аналитический метод исследования, основанный на масс-спектрометрическом измерении. Внедрен во многих странах, для проведения неонатального скрининга начал применяться и в России. Метод позволяет одновременно определять уровень тирозина и соотношение фенилаланин/тирозин.
Биологическим материалом для исследования служат высушенные пятна капиллярной крови новорожденных на фильтровальной бумаге. Главным критерием диагностики ГФА является повышенное содержание фенилаланина в крови, нормальный уровень которого в крови у здоровых людей составляет 0-2 мг/дл.
В родильном доме у всех новорожденных на 4-й день жизни (у недоношенных на 7-й день) берется кровь из пятки на тест-бланки, которые доставляются в лабораторию медико-генетической консультации, осуществляющей определение содержания ФА в крови. Значение фенилаланина выше 2,0 мг/дл классифицируется как ГФА, которая требует проведения уточняющей диагностики.
ГФА может быть обусловлена классической (типичной) ФКУ, связанной с недостаточностью фенилаланингидроксилазы, птерин-зависимыми (кофакторными) формами болезни, резистентными к диетотерапии, наследственной гиперфенилаланинемией (доброкачественной ГФА), другими формами нарушения метаболизма (тирозинемия, галактоземия и др.). На уточняющем этапе проводится повторное обследование всех детей с первичной гиперфенилаланинемией. При содержании ФА в крови от 2,1 до 8,0 мг/дл предполагается доброкачественная ГФА.
Ребенок наблюдается в медико-генетической консультации в течение первого года жизни с ежемесячным контролем уровня ФА крови. При концентрации ФА в крови выше 8,0 мг/дл диагностируется фенилкетонурия, назначается диетотерапия, на основании эффективности которой планируются мероприятия по уточнению диагноза и выбору дальнейшей тактики необходимого лечения.
Для уточнения нозологической формы ГФА в некоторых странах проводится фенилаланин-нагрузочный тест с определением концентрации тирозина и активности ФАГ. Повышенная концентрация тирозина в нагрузочном тесте свидетельствует о доброкачественном или транзиторном характере ГФА.
Необходимо учитывать, что при определении активности 13 ФАГ в половине случаев классической ФКУ обнаруживается остаточная активность ФАГ, составляющая до 6% от нормы, что связано с изменением вторичной структуры фермента вследствие однонуклеотидных замен и точковых делеций в гене.
В РФ данный метод не применяется.
Следующим этапом для уточнения классической фенилкетонурии является молекулярно-генетическая диагностика. В большинстве лабораторий существуют наборы, позволяющие определять частые мутации в гене ФАГ, имеющиеся у 80% больных ФКУ.
- При отсутствии исследуемых мутаций у пациента рекомендуется проведение секвенирования гена РАН.
- Для исключения птерин-зависимых форм ФКУ во многих развитых странах у лиц с гиперфенилаланинемией исследуются птерины в моче.
- В РФ при подозрении на данные формы диагноз подтверждается молекулярно- генетическим методом.
Следует отметить, что при определенных мутациях в гене РАН при введении кофактора BH4 активность фермента ФАГ восстанавливается, в таком случае соблюдение диеты не требуется или диета расширяется с увеличением в рационе белковых продуктов.
При подозрении на BH4- чувствительную форму в ряде стран проводится нагрузка с ВН4 при употреблении белковых продуктов под контролем уровня фенилаланина в крови.
Отсутствие нарастания уровня ФА в крови позволяет подтвердить данную форму патологии.
На заключительном этапе проводится медико-генетическое консультирование семьи, планируется пренатальная диагностика.
При отсутствии данных неонатального скрининга диагностика заболевания основывается на совокупности генеалогического анамнеза, результатов клинического и биохимического обследования, возможна молекулярная диагностика.
Главным биохимическим критерием для установления диагноза остается высокое содержание фенилаланина в сыворотке крови.

(классификация
дана по Mc
Kusik
V.A.,
1988)
Классическая
фенилкетонурия (ФКУ) описана А.
Folling.,1934
г.
Заболевание
наследуется
аутосомно-рецессивно
и вызвано мутацией гена, локализующегося
в длинном плече 12 хромосомы.
В
основе
болезни
лежит дефицит фермента фенилаланин -4-
гидроксилазы, обеспечивающего превращение
фенилаланина в тирозин. В результате
метаболического блока происходит
значительное накопление в тканях и
жидкостях больного организма фенилаланина
и таких его производных, как
фенилпировиноградная, фенилмолочная,
фенилуксусная кислоты, фенилэтиламин,
фенилацетилглютамин,
и др.
В
патогенезе ФКУ
имеют значение следующие механизмы:
Прямое токсическое действие на ЦНС фенилаланина и его производных;
Нарушение в обмене белков, липо- и гликопротеидов;
Расстройства транспорта аминокислот;
Нарушение метаболизма гормонов;
Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
Нарушение функции печени : диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота
классической
ФКУ среди новорожденных по данным
массового скрининга в среднем колеблется
от 1 : 5000 до 1 : 10000 по разным регионам
России.
Впервые
атипичная ФКУ описана I.
Smith,
1974 г.
Заболевание связано с дефицитом
дигидроптеридинредуктазы.
Заболевание
наследуется аутосомно-рецессивно.
Генный дефект локализуется в коротком
плече 4 хромосомы, участке 4р 15.3.
В
результате недостаточности
дигидроптеридинредуктазы нарушается
восстановление активной формы
тетрагидробиоптерина, участвующего в
качестве кофактора в гидроксилировании
фенилаланина, тирозина, и триптофана.
Частота
заболевания
составляет 1 : 100 000 новорожденных.
Рано
начатое лечение способствует нормализации
фенилаланина в крови, однако не
предупреждает появление клинической
симптоматики, которая развивается в
начале второго полугодия жизни.
Фенилкетонурию 2 называют диеторезистентной
ФКУ.
Этот
вариант болезни описал S.
Kaufman
в
1978 г. Заболевание наследуется
аутосомно-рецессивно и связано с
недостаточностью 6-пирувоилтетрагидроптерин
синтетазы, участвующей в процессе
синтеза тетрагидробиоптерина.
Развивающиеся при этом расстройства
сходны с нарушениями, наблюдаемыми при
ФКУ 2.
- Частота
болезни
составляет 1 : 30 000 новорожденных. - Фенилкетонурия
3 также диеторезистентна - ДРУГИЕ
ВАРИАНТЫ ФКУ.
Эти
формы ФКУ связаны с нарушением
альтернативных путей обмена фенилаланина.
Формируется метилминдальная
ацидурия и парагидроскифенилуксусная
ацидурия.
Заболевание
развивается у потомков женщин, страдающих
ФКУ и не получающих диету в зрелом
возрасте. Патогенез
мало
изучен, предполагается, что он сходен
с патогенезом остальных форм ФКУ.
Тяжесть
поражения плода коррелирует с уровнем
фенилаланина в плазме матери. Так
как эмбрион особенно чувствителен
к тератогенным воздействиям , рекомендуется
начинать диету еще до наступления
беременности.
В суточном рационе
использовать менее 15-20 мг/кг
фенилаланина. При это важно избегать
дефицита незаменимых аминокислот.
При
рождении больные фенилкетонурией не
отличаются от других новорожденных.
Манифестация ФКУ происходит обычно в
возрасте 2-6 месяцев.
Первыми
проявлениями болезни
служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- срыгивание, рвота;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
- судорожный синдром;
- заплесневелый, мышиный, волчий запах мочи и пота.
- При
отсутствии лечения формируется задержка
статико-моторного и психоречевого
развития, умственная отсталость
достигает, как правило, глубокой
степени. - Характерны
такие фенотипические особенности, как
экзематозные и себорейные сыпи,
гипопигментация кожи, волос, радужной
оболочки глаз. - ДИАГНОСТИКА
ФЕНИЛКЕТОНУРИИ. - ФКУ
может быть диагностирована на основе
обнаружения следующих признаков:
- стойкой гиперфенилаланинемии (более 240 ммоль/л);
- вторичного дефицита тирозина;
- экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
В
настоящее время, согласно приказу
Минздрава России № 316 от 30.12.93 проведение
неонатального скрининга на ФКУ стало
обязательным. Скрининирующие тесты
должны быть простыми, недорогими и
информативными. Этим требованиям
отвечают методы, используемые для
ранней диагностики ФКУ:
- микробиологический тест Гатри;
- метод флюоресцирующих антител (лабораторный комплекс “Флюороскан”, позволяющий проводить 800 проб в час);
- метод тонкослойной хроматографии .
- Оптимальные
сроки обследования новорожденных —
доношенных, зрелых — 5-6 день жизни;
недоношенных, незрелых, больных —
10-14 день жизни. - Трактовка результатов:
- 1 группа
— уровень фенилаланина не превышает 200
ммоль/л
(1-3 мг%)
— норма;
2 группа
— уровень фенилаланина составляет
200-500
ммоль/л
(3-10 мг%) — гиперфенилаланинемия. В
эту группу входят дети с транзиторной
гиперфенилаланинемией, вследствие
незрелости ферментных систем печени и
больные ФКУ.
За данной группой проводится
наблюдение по следующему плану: если в
течение 6 недель при еженедельном
исследовании уровень фенилаланина
остается менее 500 ммоль/л, контроль за
уровнем фенилаланина крови проводят
до 1 года первоначально каждые 3 месяца,
а затем каждые 6 мес. жизни.
При уровне
более 500 ммоль/л назначается диетотерапия.
3 группа
— уровень фенилаланина превышает 500
ммоль/л
(более 10 мг%), диагностируется ФКУ и с
момента постановки диагноза назначается
диетотерапия.
В
настоящее время разрабатываются и
внедряются молекулярно-генетические
методы диагностики генного дефекта при
ФКУ. Прямая диагностика мутантного гена
проводится с помощью синтетических
олигонуклеотидных зондов, этот метод
пригоден для дородовой диагностики ФКУ
и выявления гетерозиготного носительства.
Помимо
молекулярно-генетического анализа,
выявление гетерозигот может осуществляться
биохимическими тестами после нагрузки
фенилаланином в дозе 25 мг/кг.
Диеторезистентные
формы ФКУ диагностируют при помощи:
- исследования биоптеринов мочи ;
- перорального нагрузочного теста с тетрагидробоиптерином (через 4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит резкое снижение и нормализация уровня фенилаланина в крови с одновременным повышением уровня тирозина);
- исследования активности дигидроптеридинредуктазы и 6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов, эритроцитах, гепатоцитах.
Главным способом лечения является диетотерапия, ограничивающая поступление в организм фенилаланина; приступить к ней нужно немедленно после установления диагноза.
Препараты с промедиаторным действием:
Диеторезистентные формы. Лечение включает назначение тетрагидробиоптерина — доза 10-20 мг/кг в сутки.
- Выявление гетерозиготных носителей;
- Внедрение программ массового скрининга новорожденных для раннего выявления ФКУ и своевременного назначения диетотерапии;
- Пренатальная диагностика.
источник